Pituitary Gland Disorders
This group of endocrine diseases includes the following conditions
- Hypopituitarism (global pituitary hormone deficiency)
- Prolactinoma (already covered under reproductive disorders)
- Cushing's Disease (Cushing's Syndrome discussed under Adrenal Diseases)
- Craniopharyngioma
- Pituitary Apoplexy
- Empty Sella Syndrome
- Diabetes Insipidus (Central)
- Syndrome of Inappropriate ADH Secretion (SIADH)
1. Hypopituitarism (global pituitary hormone deficiency)
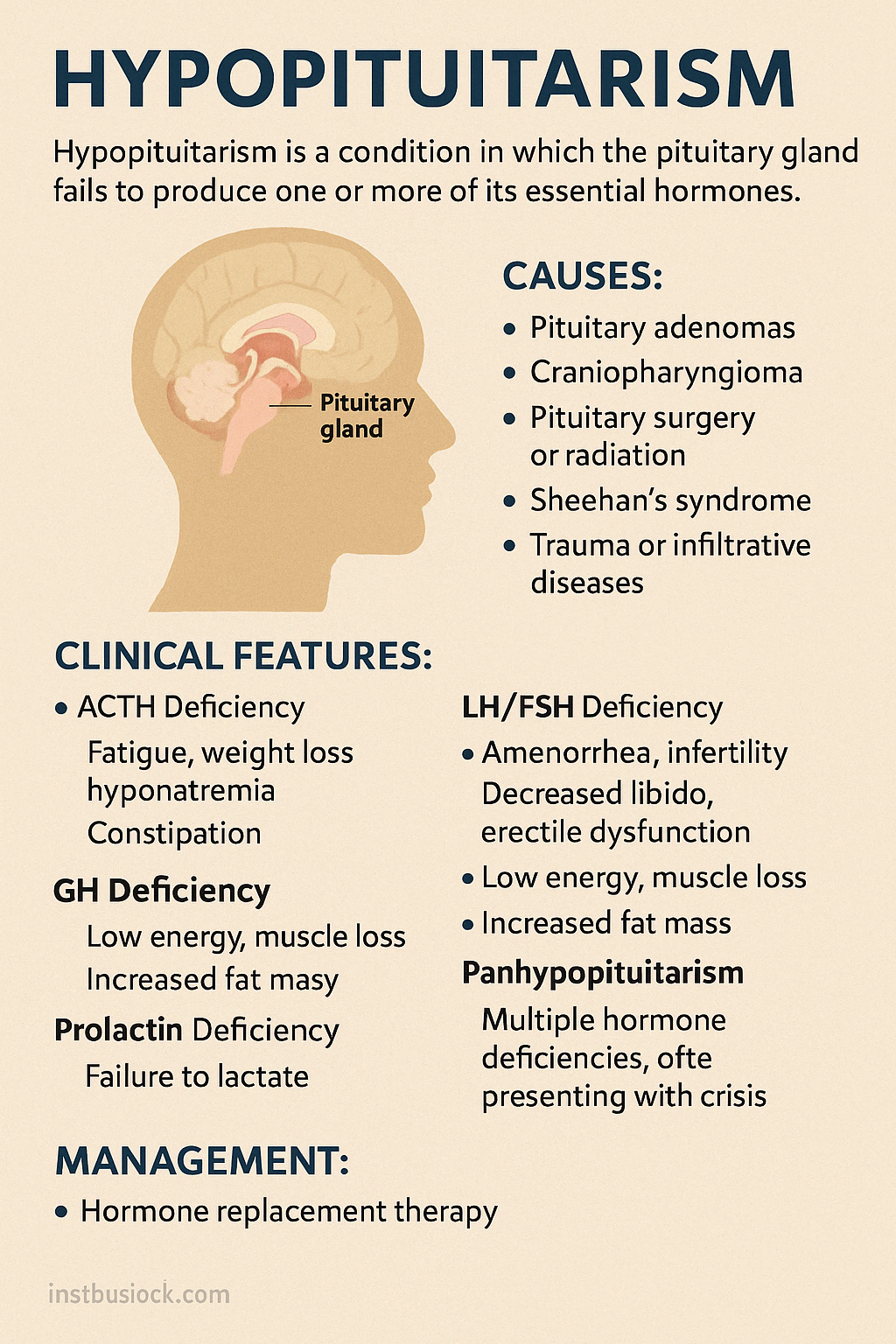
Hypopituitarism is a condition in which the pituitary gland fails to produce one or more of its essential hormones, leading to partial or complete deficiency of downstream endocrine functions. It may affect secretion of ACTH, TSH, LH, FSH, GH, and prolactin, depending on the cause and extent of damage.
Causes:
- Pituitary adenomas (non-functioning tumors)
- Craniopharyngioma or other parasellar masses
- Pituitary surgery or radiation
- Sheehan's syndrome (postpartum pituitary infarction)
- Trauma, infections, infiltrative diseases (e.g., sarcoidosis)
- Genetic defects (congenital hypopituitarism)
Clinical Features:
Symptoms vary based on which hormones are deficient:
- ACTH deficiency: fatigue, weight loss, low blood pressure, hyponatremia
- TSH deficiency: cold intolerance, bradycardia, constipation
- LH/FSH deficiency: amenorrhea, infertility, decreased libido, erectile dysfunction
- GH deficiency (in adults): low energy, muscle loss, increased fat mass
- Prolactin deficiency: failure to lactate postpartum
- Panhypopituitarism: multiple hormone deficiencies, often presenting with crisis
In children, it may cause growth failure, delayed puberty, and developmental delays.
Diagnosis:
- Hormone testing: low target gland hormones with inappropriately low pituitary hormones (e.g., low cortisol and ACTH, low T4 and TSH)
- MRI of the pituitary to assess structural abnormalities
Management:
- Hormone replacement therapy: glucocorticoids, levothyroxine, sex steroids, GH as needed
- Replace hydrocortisone before thyroxine to prevent adrenal crisis
- Lifelong treatment and regular monitoring required
With appropriate hormonal therapy and monitoring, most individuals with hypopituitarism can lead healthy and functional lives.
2. Prolactinoma (already covered under reproductive disorders)
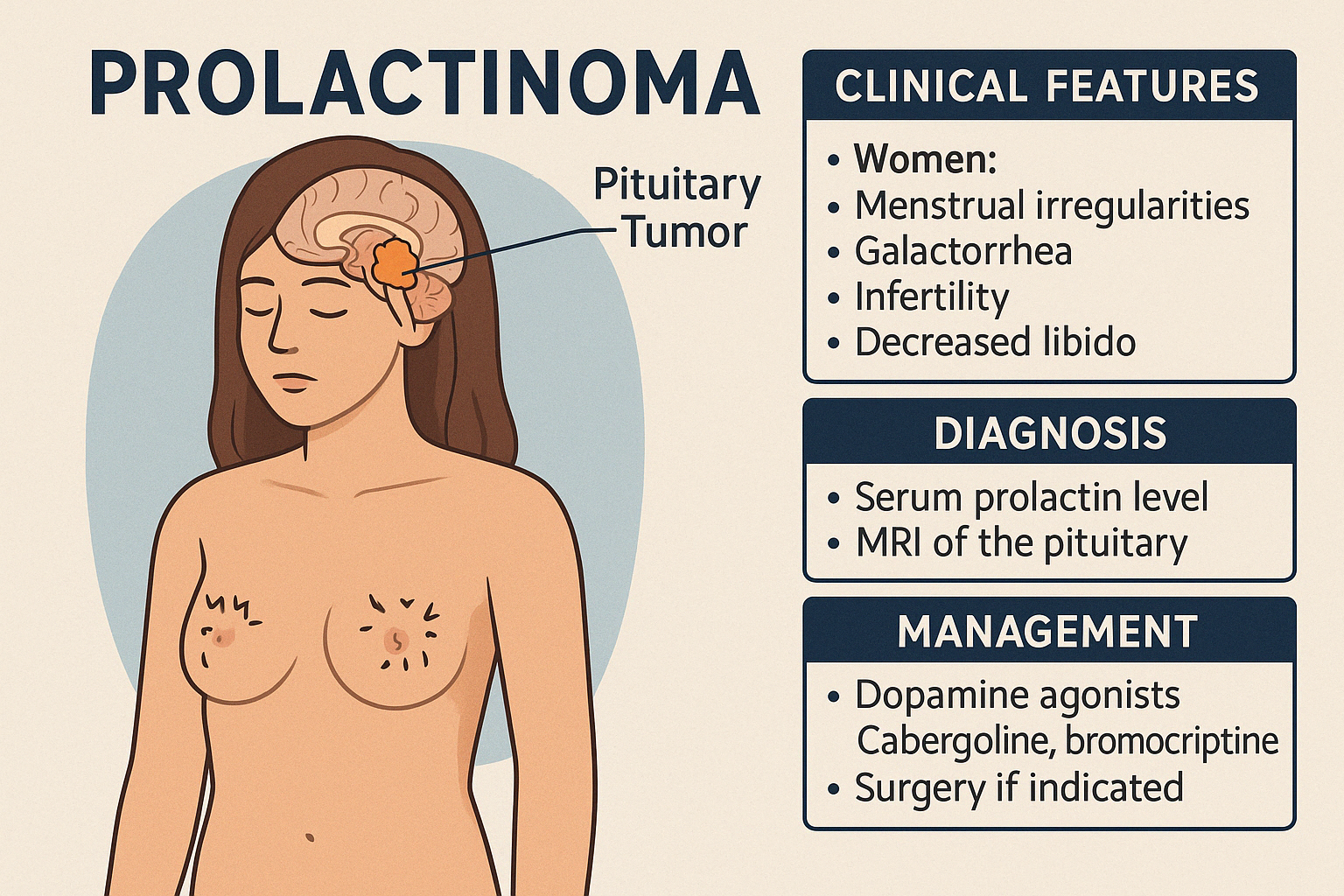
A Prolactinoma is a benign pituitary adenoma that produces excessive prolactin, the hormone responsible for lactation. It is the most common type of functioning pituitary tumor and occurs more frequently in women of reproductive age, though it also affects men.
Pathophysiology
The overproduction of prolactin inhibits GnRH secretion, leading to suppressed LH and FSH, and subsequently reduced estrogen or testosterone levels, affecting reproductive function.
Prolactinomas are classified by size:
- Microadenomas (<1 cm)
- Macroadenomas (≥1 cm), which may compress surrounding structures, especially the optic chiasm
Clinical Features
In women:
- Menstrual irregularities or amenorrhea
- Galactorrhea (milk discharge unrelated to pregnancy)
- Infertility
- Decreased libido
In men:
- Decreased libido, erectile dysfunction
- Infertility
- Gynecomastia
- Occasionally galactorrhea
- Headache or visual field defects (if macroadenoma)
Diagnosis
- Serum prolactin level (markedly elevated)
- MRI of the pituitary
- Rule out secondary causes (e.g., hypothyroidism, medications)
Management
- Dopamine agonists (first-line):
- Cabergoline (preferred)
- Bromocriptine
- These medications often reduce prolactin levels, shrink the tumor, and restore fertility
- Surgery (transsphenoidal) if medical therapy fails or rapid visual deterioration occurs
- Lifelong follow-up is usually required
Prolactinomas are one of the most treatable pituitary tumors, with excellent outcomes when managed appropriately.
3. Cushing's Disease (Cushing's Syndrome discussed under Adrenal Diseases)
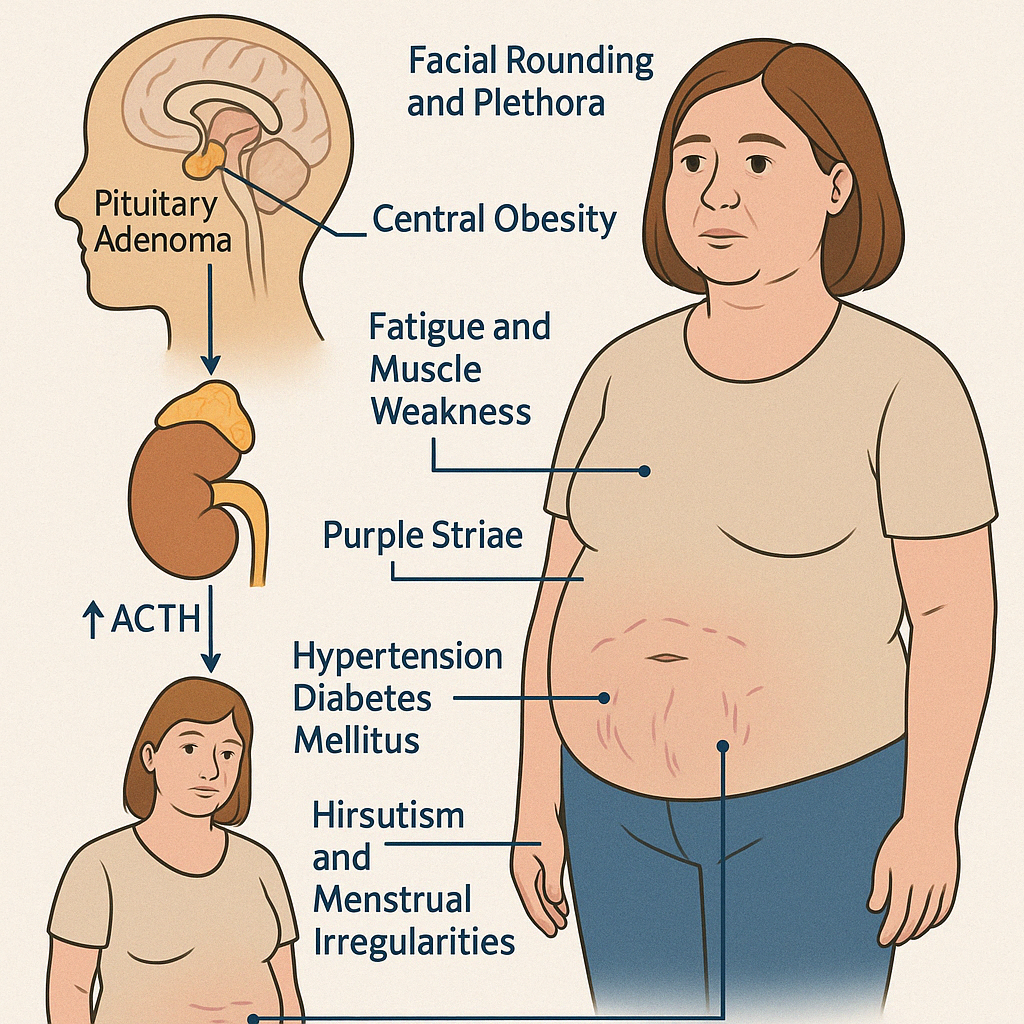
Cushing's Disease refers specifically to excess production of adrenocorticotropic hormone (ACTH) by a pituitary adenoma, leading to overstimulation of the adrenal glands and subsequent hypercortisolism. It is the most common cause of endogenous Cushing's Syndrome.
Clinical Features:
- Central obesity (moon face, dorsocervical fat pad, abdominal fat)
- Facial rounding and plethora
- Purple striae (particularly on the abdomen and thighs)
- Muscle weakness, especially proximal limbs
- Hypertension, diabetes mellitus, and dyslipidemia
- Hirsutism, acne, and menstrual irregularities in women
- Depression, emotional lability, or cognitive impairment
- Osteopenia or osteoporosis
- Easy bruising, poor wound healing
- In children: growth retardation with weight gain
Diagnosis:
1. Screening Tests:
- 24-hour urinary free cortisol (UFC)
- Low-dose dexamethasone suppression test
- Late-night salivary cortisol
2. Confirmatory Tests:
- Elevated plasma ACTH
- Lack of cortisol suppression on dexamethasone testing
3. Localization:
- MRI of the pituitary
- High-dose dexamethasone suppression (suppression seen in pituitary causes)
- Inferior petrosal sinus sampling (IPSS) when imaging is inconclusive
Management:
- Transsphenoidal surgery is the first-line treatment
- Medical therapy if surgery is not curative:
- Ketoconazole, Metyrapone, Osilodrostat to inhibit cortisol synthesis
- Pasireotide for pituitary-directed therapy
- Radiation therapy or bilateral adrenalectomy in refractory cases
- Hormone replacement post-surgery if hypopituitarism develops
Engage With Dr. Vipin Mishra
Whether you are dealing with chronic endocrine condition or just want to understand your body better, or wish to obtain an exalted consciousness, you can take help and guidance from Dr. Vipin Mishra.
Engage With Dr. Vipin Mishra4. Craniopharyngioma
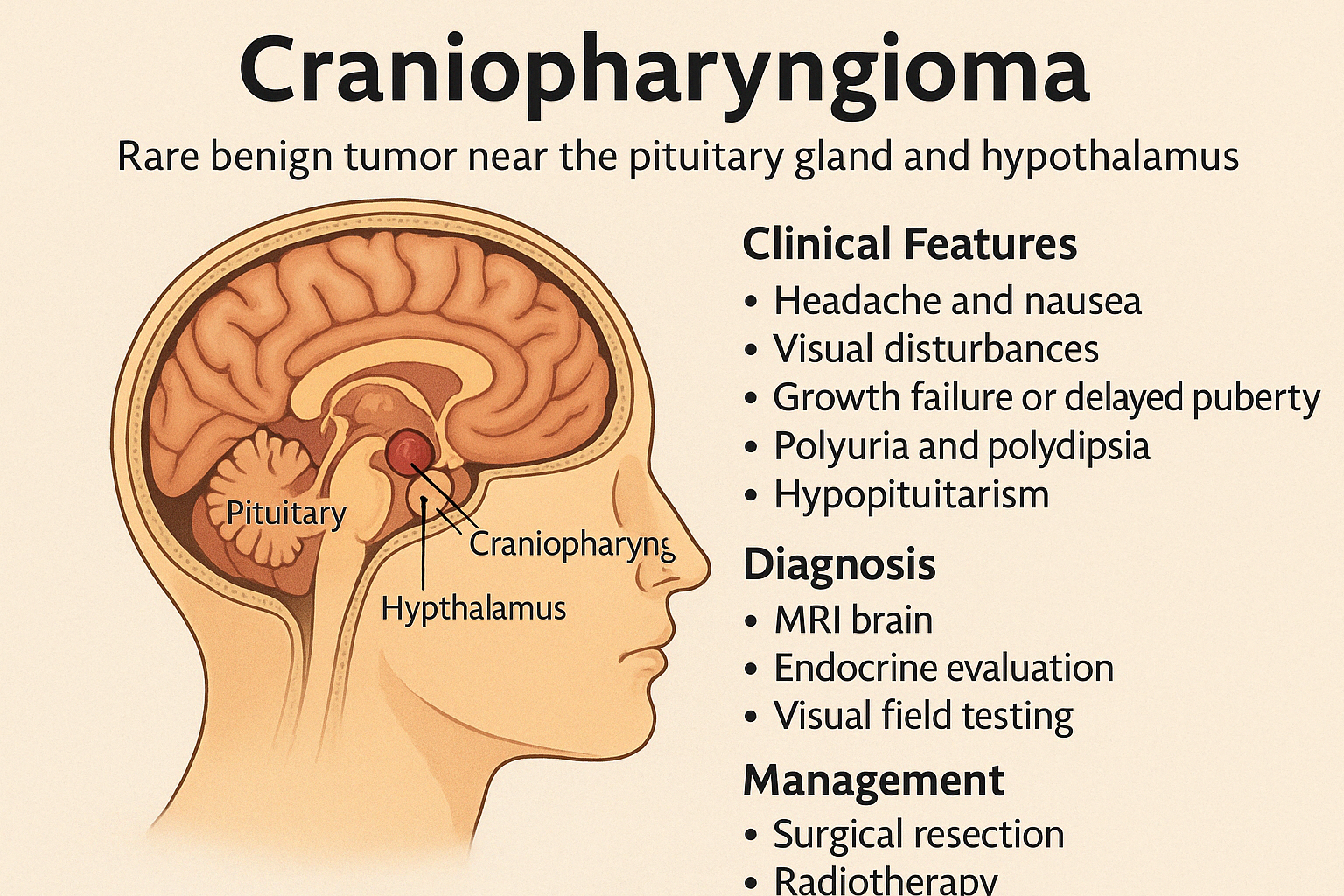
Craniopharyngioma is a rare, benign but locally aggressive brain tumor that arises near the pituitary gland and hypothalamus, often from remnants of Rathke's pouch. Despite being histologically benign, it can cause significant endocrine and neurological problemsdue to its location.
Epidemiology
Bimodal age distribution:
- Children (5–14 years)
- Adults (50–70 years)
Clinical Features
Due to compression of surrounding structures, patients may present with:
- Headache and nausea (raised intracranial pressure)
- Visual disturbances (bitemporal hemianopia from optic chiasm compression)
- Growth failure or delayed puberty (due to GH and gonadotropin deficiency)
- Polyuria and polydipsia (from hypothalamic damage → central diabetes insipidus)
- Obesity and behavioral changes (hypothalamic involvement)
- Hypopituitarism (multiple hormone deficiencies)
In children, growth failure and delayed puberty are often the first signs.
Diagnosis
- MRI brain: reveals a cystic/solid mass with calcifications near the pituitary-hypothalamic region
- Endocrine evaluation: GH, TSH, ACTH, LH/FSH, prolactin, cortisol, and thyroid levels
- Visual field testing
Management
- Surgical resection is the primary treatment (total or subtotal)
- Radiotherapy often follows partial resection or for recurrent tumors
- Hormone replacement therapy is essential postoperatively for endocrine deficits
- Lifelong follow-up is needed for endocrine, neurological, and psychological monitoring
Early diagnosis and a multidisciplinary approach can greatly improve outcomes despite the complexity of this tumor.
5. Pituitary Apoplexy
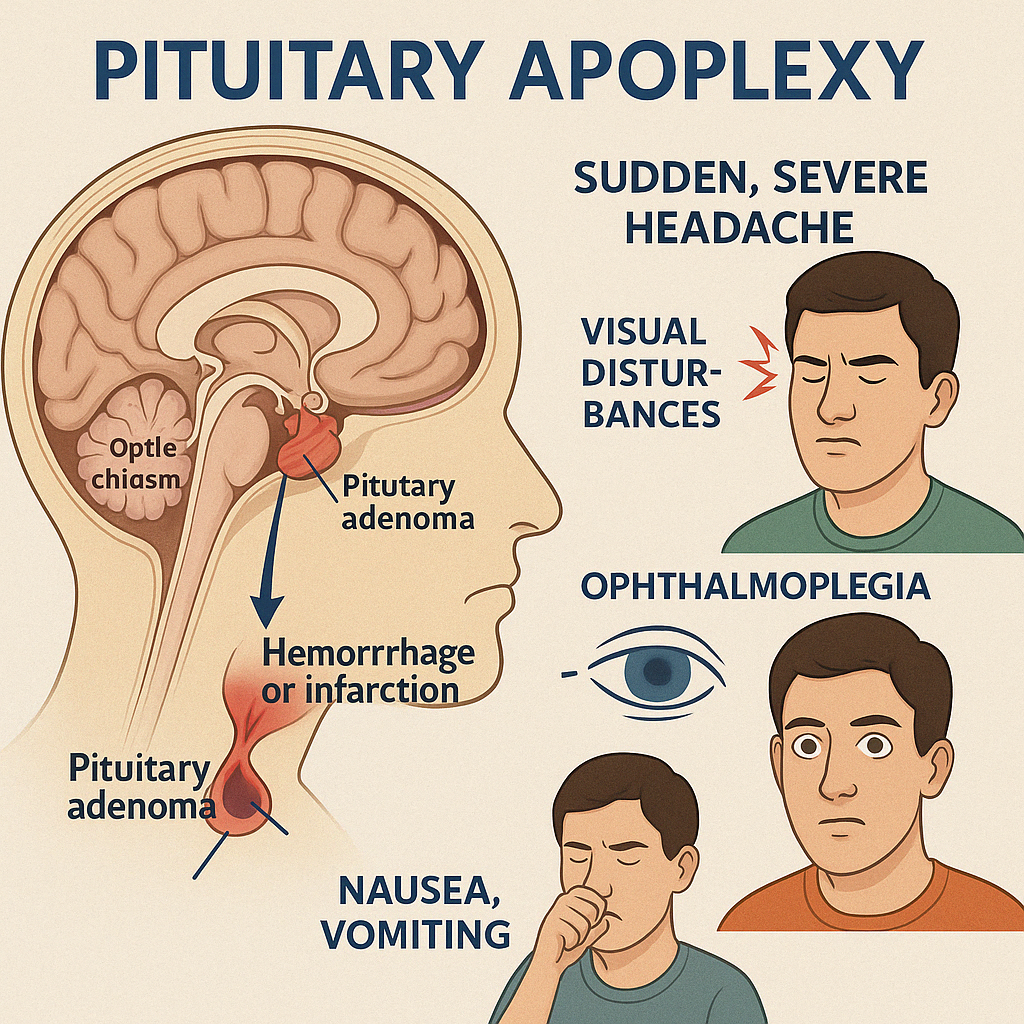
Pituitary Apoplexy is a rare, potentially life-threatening endocrine emergency caused by sudden hemorrhage or infarction within the pituitary gland, most often in a pre-existing pituitary adenoma. The abrupt expansion of the gland causes pressure on surrounding structures and acute hormone deficiencies, particularly cortisol.
Causes and Risk Factors
- Pituitary macroadenoma (often previously undiagnosed)
- Hypertension
- Major surgery or trauma
- Anticoagulant therapy
- Dynamic pituitary testing or pregnancy (very rare)
Clinical Presentation
- Sudden, severe headache (often retro-orbital)
- Visual disturbances: decreased visual acuity or bitemporal hemianopia
- Ophthalmoplegia: due to cranial nerve compression (III, IV, VI)
- Nausea, vomiting
- Altered consciousness (if severe)
- Signs of acute adrenal insufficiency: hypotension, hyponatremia, shock
Diagnosis
- Urgent MRI or CT scan: shows hemorrhage or necrosis within the pituitary
- Hormonal evaluation: low cortisol, ACTH, TSH, LH/FSH, prolactin
- Visual field testing if conscious and cooperative
Management
- Immediate intravenous hydrocortisone is lifesaving
- Supportive care for blood pressure and electrolytes
- Neurosurgical decompression if visual or neurological deficits progress
- Long-term hormone replacement often required (hydrocortisone, thyroxine, sex hormones ± GH)
Early recognition and prompt treatment are essential to prevent mortality and long-term complications.
6. Empty Sella Syndrome
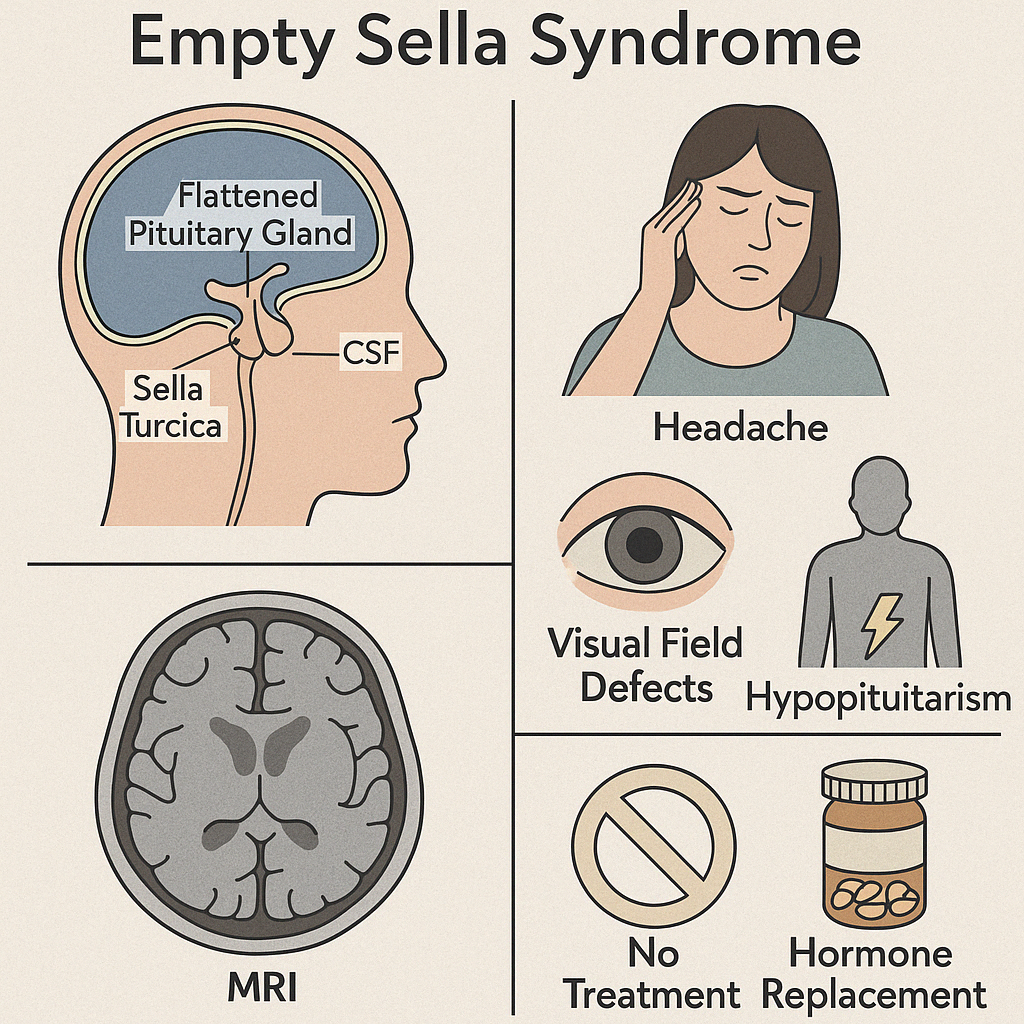
Empty Sella Syndrome (ESS) refers to a radiological finding where the sella turcica, the bony cavity that normally houses the pituitary gland, appears partially or completely filled with cerebrospinal fluid (CSF) — making the pituitary gland look flattened or "absent."
It is not always associated with hormonal dysfunction but may occur with pituitary hypofunction, visual symptoms, or as an incidental finding.
Types:
1. Primary ESS
- Often due to a congenital defect in the diaphragma sellae
- More common in middle-aged, obese women with hypertension
- Sometimes linked with idiopathic intracranial hypertension
2. Secondary ESS
- Occurs after pituitary surgery, radiation, infarction, or apoplexy
- Associated with loss of pituitary tissue
Clinical Features:
- Often asymptomatic (incidental finding on brain imaging)
- May present with:
- Headache
- Visual field defects (if optic chiasm is herniated)
- Signs of hypopituitarism: fatigue, menstrual irregularity, low libido
- CSF rhinorrhea (rare)
Diagnosis:
- MRI of the brain: shows CSF filling the sella with a thin rim of pituitary tissue
- Hormonal testing to assess pituitary function (TSH, cortisol, LH/FSH, IGF-1, prolactin)
Management:
- No treatment if asymptomatic and pituitary function is intact
- Hormone replacement therapy if deficiencies are found
- Surgical repair in rare cases with CSF leak or optic chiasm herniation
Most patients with ESS live normal lives, especially when identified early and monitored for evolving hormonal deficits.
7. Diabetes Insipidus (Central)
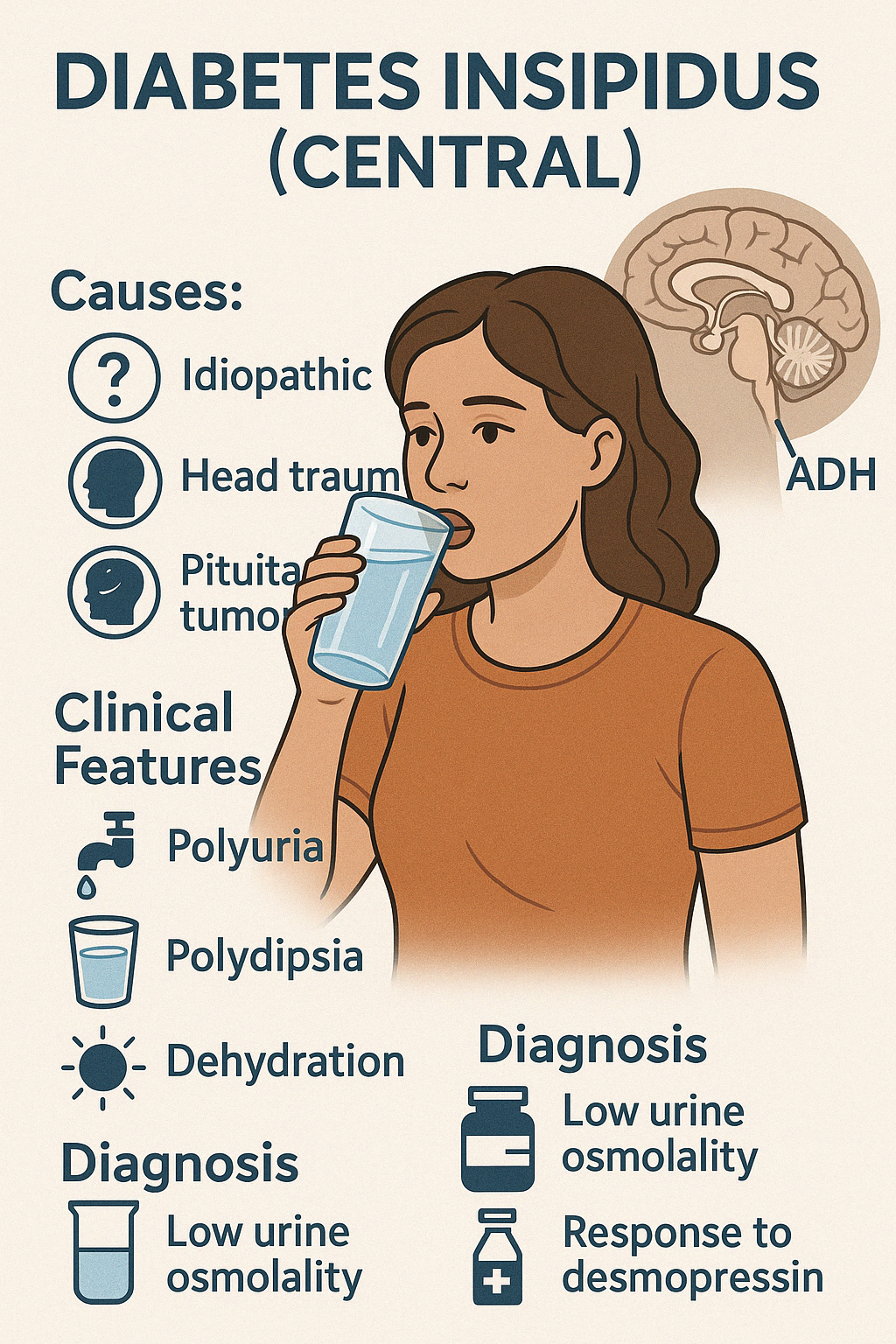
Central Diabetes Insipidus (DI) is a disorder characterized by deficient secretion of antidiuretic hormone (ADH), also called vasopressin, from the posterior pituitary. ADH normally regulates water reabsorption in the kidneys. In its absence, the kidneys are unable to concentrate urine, leading to excessive water loss.
Causes:
- Idiopathic (most common)
- Trauma or neurosurgery involving the pituitary or hypothalamus
- Pituitary tumors or craniopharyngioma
- Infections (e.g., encephalitis, meningitis)
- Autoimmune disorders or infiltrative diseases (e.g., Langerhans cell histiocytosis, sarcoidosis)
Clinical Features:
- Polyuria (large volumes of dilute urine, >3–4 liters/day in adults)
- Polydipsia (especially for cold water)
- Nocturia
- If water intake is inadequate, dehydration, hypotension, and hypernatremia may occur
- In infants and elderly, symptoms may be more subtle but dangerous
Diagnosis:
- Low urine osmolality despite high serum osmolality
- Water deprivation test: failure to concentrate urine
- Response to desmopressin (synthetic ADH) confirms central DI
- MRI brain to assess posterior pituitary "bright spot" and underlying lesions
Management:
- Desmopressin (DDAVP): intranasal, oral, or injectable forms
- Ensure adequate fluid intake
- Monitor for electrolyte imbalances, especially hyponatremia if overcorrected
- Treat underlying cause when identifiable
With appropriate treatment, patients with central DI can live normal lives with minimal restrictions.
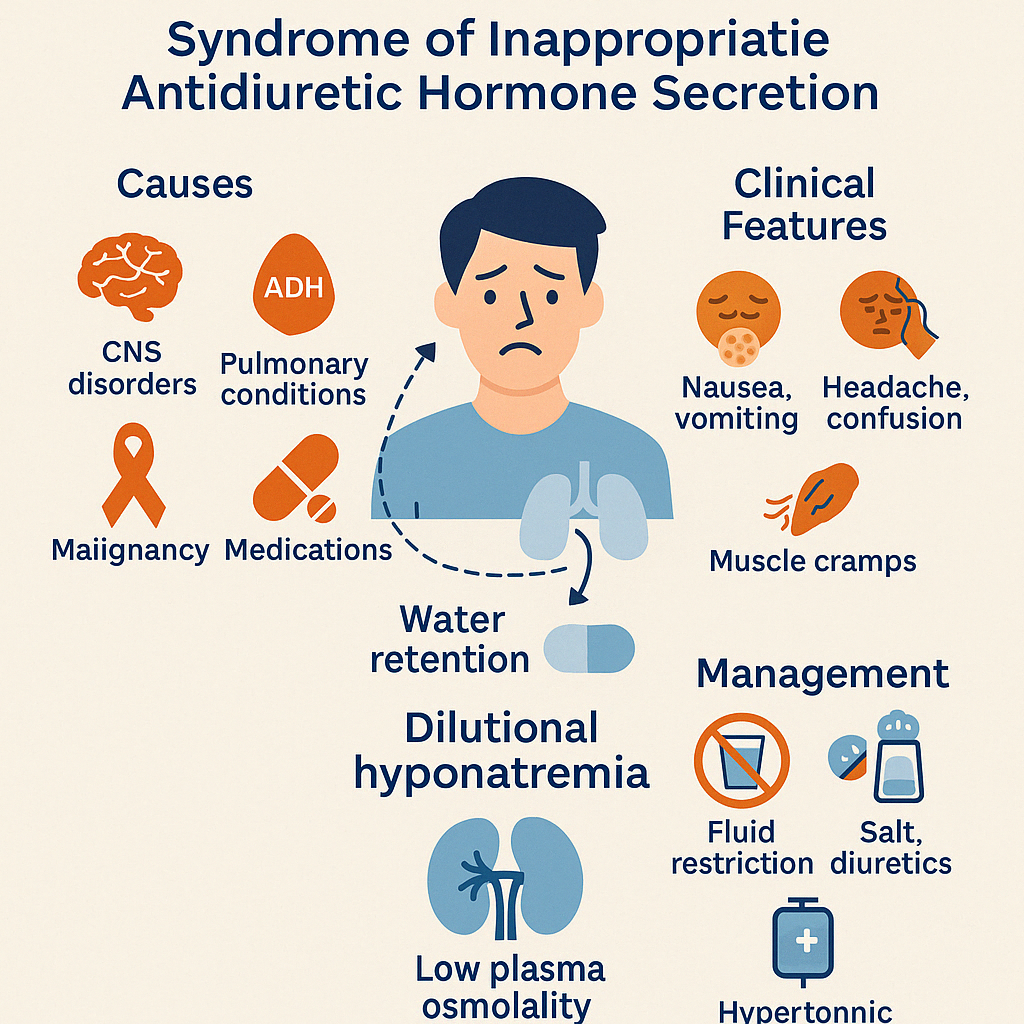
8. Syndrome of Inappropriate ADH Secretion (SIADH)
SIADH is a condition characterized by excessive release of antidiuretic hormone (ADH) despite normal or low plasma osmolality, leading to water retention, dilutional hyponatremia, and concentrated urine.
Unlike in diabetes insipidus (where ADH is deficient), SIADH results in too much ADH, causing the kidneys to retain water and suppress aldosterone, which dilutes the blood sodium level.
Causes:
- CNS disorders: stroke, hemorrhage, trauma, meningitis, tumors
- Pulmonary conditions: pneumonia, tuberculosis, mechanical ventilation
- Malignancies (especially small-cell lung carcinoma)
- Medications: SSRIs, carbamazepine, cyclophosphamide, antipsychotics
- Postoperative state or pain
Clinical Features:
- Symptoms of hyponatremia:
- Nausea, vomiting
- Headache, confusion
- Muscle cramps
- Seizures, coma (in severe cases)
- No edema (euvolemic state) despite fluid retention
Diagnosis:
- Low serum sodium (<135 mmol/L)
- Low plasma osmolality
- Inappropriately concentrated urine (high urine osmolality)
- Urine sodium > 40 mmol/L
- Normal renal, adrenal, and thyroid function
Management:
- Fluid restriction (mainstay of treatment)
- Salt tablets and loop diuretics in some cases
- Hypertonic saline in symptomatic or severe hyponatremia
- Vaptans (ADH receptor antagonists) for resistant cases
- Address underlying cause (e.g., infection, tumor, medication)
Careful correction of sodium is essential to avoid osmotic demyelination syndrome. SIADH requires a nuanced and monitored approach for optimal outcomes.