Genetic and Congenital Endocrine Conditions
This group of endocrine diseases includes the following conditions
- Congenital Adrenal Hyperplasia (CAH) (already covered under adrenal diseases)
- Turner Syndrome
- Klinefelter Syndrome
- Androgen Insensitivity Syndrome (AIS)
- Kallmann Syndrome
- McCune–Albright Syndrome (already referenced in context of pseudohypoparathyroidism)
- Genetic forms of Diabetes (e.g., MODY, neonatal diabetes)
1. Congenital Adrenal Hyperplasia (CAH) (already covered under adrenal diseases)
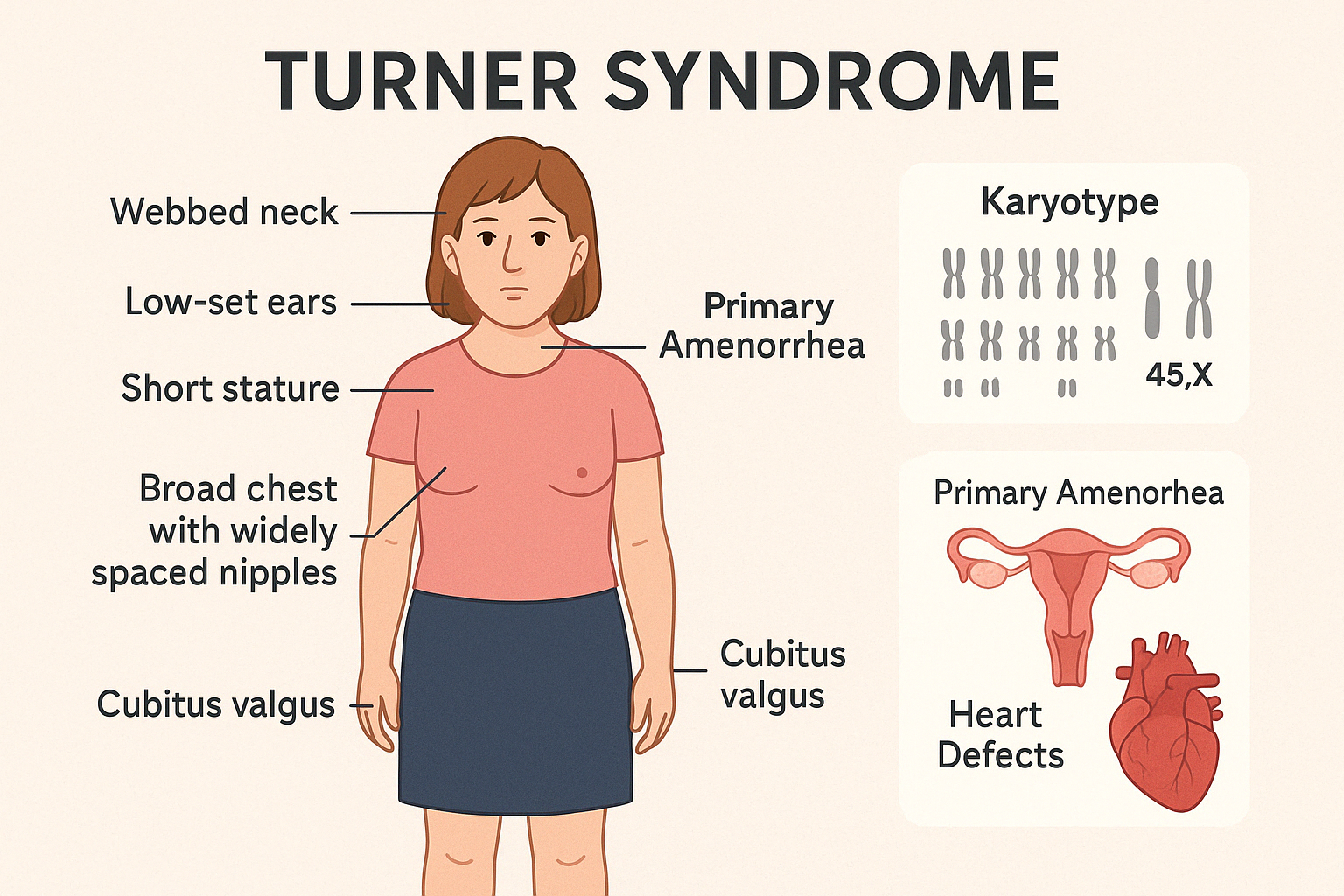
2. Turner Syndrome
Turner Syndrome is a chromosomal disorder affecting females, characterized by complete or
partial monosomy of the X chromosome (45,X or variants). It leads to a wide range of clinical
features, particularly involving growth and sexual development, along with potential
cardiovascular and endocrine complications.
Key Clinical Features:
- Short stature (often <3rd percentile)
- Primary amenorrhea and delayed puberty
- Infertility (due to gonadal dysgenesis)
- Webbed neck, low-set ears, broad chest with widely spaced nipples
- Cubitus valgus, shortened 4th metacarpals
- Congenital heart defects (especially bicuspid aortic valve and coarctation of aorta)
- Renal anomalies (e.g., horseshoe kidney)
- Normal intelligence, but may have difficulty with spatial-temporal processing
Endocrine Manifestations:
- Hypergonadotropic hypogonadism: elevated LH and FSH, low estrogen
- Hypothyroidism (increased prevalence, often autoimmune)
- Glucose intolerance, insulin resistance, and risk of type 2 diabetes
- Osteopenia/osteoporosis due to estrogen deficiency
Diagnosis:
- Karyotype analysis (45,X or mosaic forms)
- Hormonal testing for LH, FSH, estradiol
- Echocardiogram and renal ultrasound at diagnosis
- Bone age and DEXA scan in follow-up
Management:
- Growth hormone therapy to optimize final height
- Estrogen replacement therapy for pubertal induction and bone health
- Progestin added later for menstrual cycling
- Regular monitoring of cardiovascular, thyroid, and metabolic health
- Fertility counseling — oocyte donation and assisted reproduction may be considered
Early diagnosis and a multidisciplinary approach significantly enhance quality of life and long-term health outcomes.
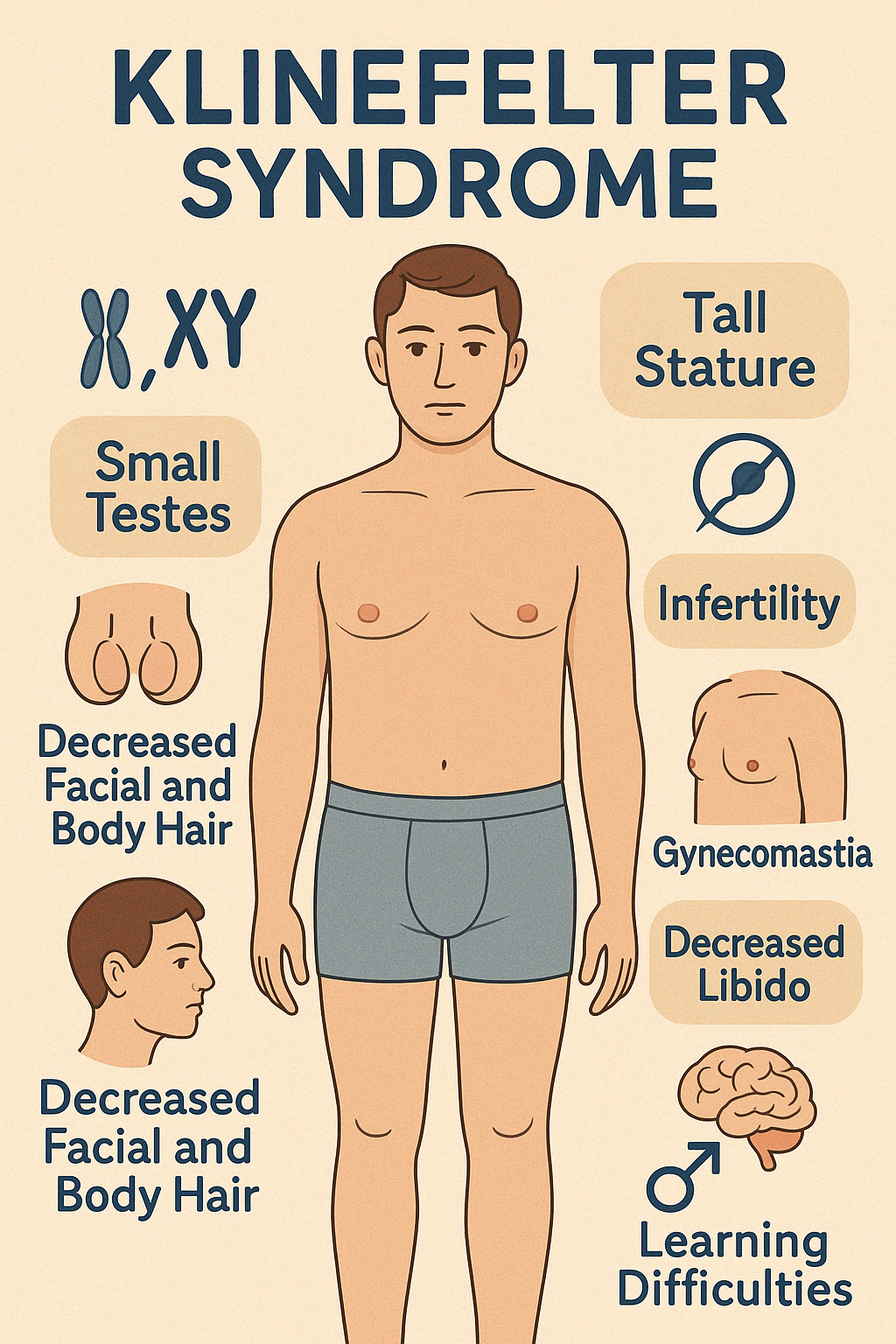
3. Klinefelter Syndrome
Klinefelter Syndrome is a chromosomal disorder in males, characterized by the presence of one
or more extra X chromosomes — most commonly 47,XXY. It is one of the most frequent causes
of male hypogonadism and infertility and often goes undiagnosed until adulthood.
Clinical Features:
- Tall stature with long limbs
- Small, firm testes (testicular atrophy)
- Gynecomastia
- Decreased facial and body hair
- Infertility due to azoospermia or oligospermia
- Low libido and erectile dysfunction
- Delayed or incomplete puberty in adolescents
- Learning difficulties (especially language and executive function)
- Mild cognitive impairment in some cases
- Increased risk of autoimmune diseases, osteoporosis, breast cancer, and metabolic syndrome
Endocrine Profile:
- Hypergonadotropic hypogonadism
- Elevated FSH and LH
- Low or low-normal testosterone
Diagnosis:
- Karyotype analysis (47,XXY or mosaic variants)
- Hormonal evaluation: testosterone, LH, FSH
- Semen analysis to assess fertility
- Bone density scan for osteoporosis
Management:
- Testosterone replacement therapy to promote secondary sexual characteristics, muscle mass, bone health, and libido
- Speech and educational support in childhood
- Fertility counseling — assisted reproduction (e.g., testicular sperm extraction with ICSI) may be possible in some
- Gynecomastia surgery if needed
- Regular follow-up for metabolic, bone, and cardiovascular health
Early diagnosis and hormone therapy can significantly improve quality of life and reduce long-term health risks.
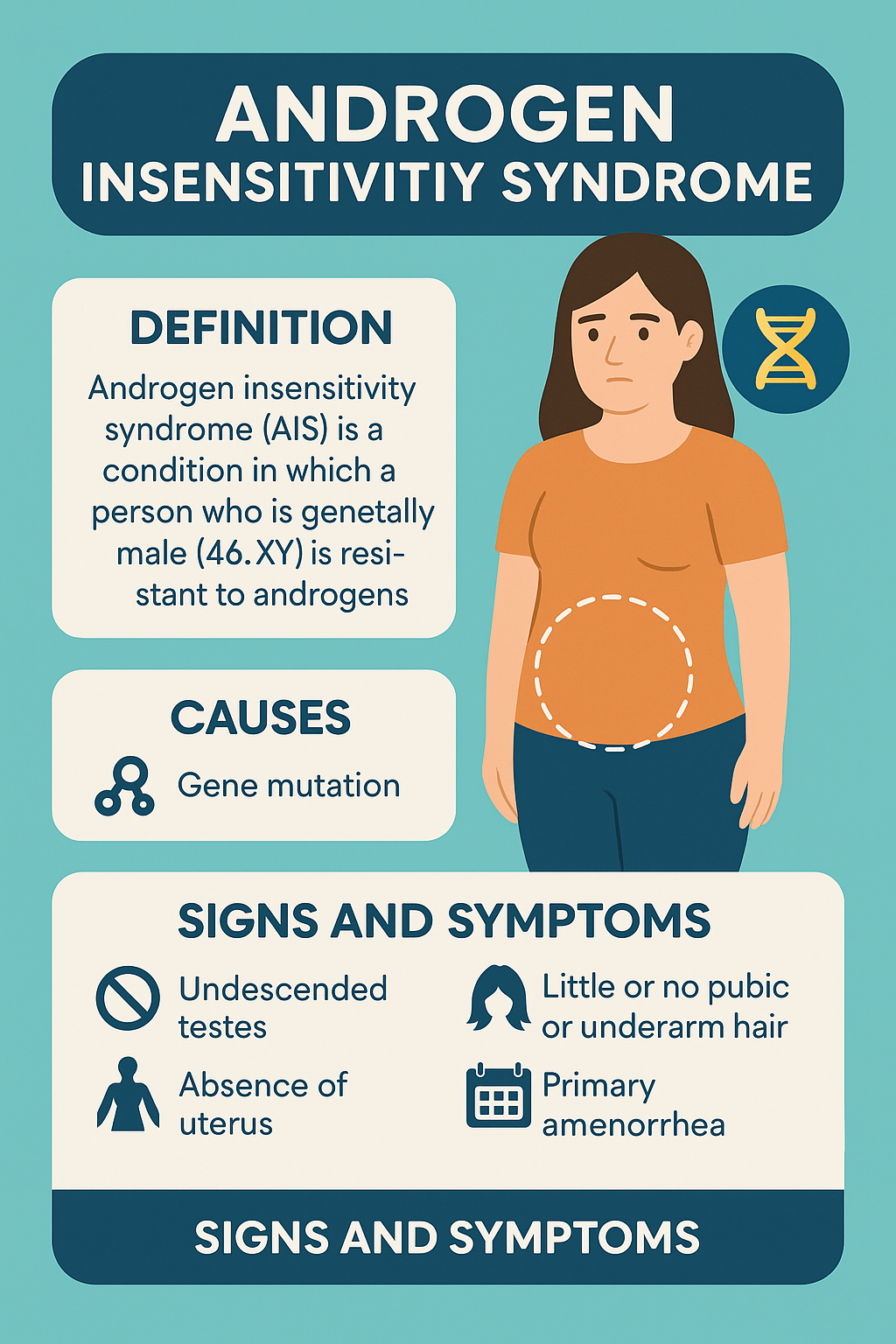
4. Androgen Insensitivity Syndrome (AIS)
Androgen Insensitivity Syndrome is a rare genetic disorder in which individuals with an XY
karyotype (genetically male) are resistant to the effects of androgens due to mutations in the
androgen receptor (AR) gene. This results in a female or ambiguous phenotype, despite the
presence of male (XY) chromosomes and testes.
AIS is classified into:
- Complete AIS (CAIS) — female external genitalia
- Partial AIS (PAIS) — ambiguous genitalia
- Mild AIS (MAIS) — undervirilized male with infertility
Clinical Features:
- In Complete AIS (CAIS):
- Phenotypic female with normal breast development
- Primary amenorrhea (no uterus, short/blind vaginal pouch)
- Absent or sparse pubic and axillary hair
- Intra-abdominal or inguinal testes (risk of malignancy later)
- Tall stature
- In Partial AIS (PAIS):
- Variable degree of genital ambiguity
- Hypospadias, micropenis, undescended testes
- Often raised as males or females depending on external appearance
- In Mild AIS (MAIS):
- Normal male genitalia but infertility, gynecomastia, or reduced virilization
Diagnosis:
- Karyotype: 46,XY
- Elevated testosterone, LH, and sometimes estradiol
- Imaging: absence of uterus and ovaries, presence of testes
- Androgen receptor gene testing confirms the diagnosis
Management:
- Gender-affirming care based on individual phenotype and preference
- Gonadectomy (after puberty in CAIS) to reduce malignancy risk
- Estrogen replacement post-gonadectomy
- Psychological support and genetic counseling
- Surgical correction or vaginal dilation therapy if needed
AIS underscores the complexity of sexual differentiation and requires a sensitive, multidisciplinary approach to care and counseling.
Engage With Dr. Vipin Mishra
Whether you are dealing with chronic endocrine condition or just want to understand your body better, or wish to obtain an exalted consciousness, you can take help and guidance from Dr. Vipin Mishra.
Engage With Dr. Vipin Mishra5. Kallmann Syndrome
Kallmann Syndrome is a genetic disorder characterized by hypogonadotropic hypogonadism
(deficient secretion of gonadotropins: LH and FSH) combined with anosmia or hyposmia
(absent or reduced sense of smell). It results from defective migration of GnRH-secreting
neurons and olfactory nerves during embryogenesis.
Etiology:
- X-linked, autosomal dominant, or autosomal recessive inheritance
- Mutations in KAL1, FGFR1, PROKR2, and other genes
- Can occur sporadically or with family history
- More common in males, but also occurs in females
Clinical Features:
- In males:
- Delayed or absent puberty
- Micropenis, cryptorchidism in infancy
- Infertility (low testosterone, azoospermia)
- Anosmia or hyposmia (often unnoticed unless tested)
- In females:
- Primary amenorrhea
- Lack of breast development
- Infertility
- Anosmia or hyposmia
- Other associated features (in some patients):
- Cleft palate, hearing loss
- Renal agenesis
- Mirror movements (synkinesia)
Diagnosis:
- Low serum LH, FSH, and sex steroids
- Normal or low GnRH (but cannot be directly measured easily)
- MRI brain: may show hypoplastic olfactory bulbs
- Smell test (e.g., UPSIT)
- Genetic testing for known mutations
Management:
- Sex hormone replacement (testosterone in males, estrogen/progesterone in females) for puberty and maintenance
- Pulsatile GnRH therapy or gonadotropin injections for fertility induction
- Psychological support and genetic counseling
- Monitoring of bone density and metabolic health
With appropriate treatment, individuals with Kallmann Syndrome can lead full and healthy lives, including achieving fertility in many cases.
6. McCune–Albright Syndrome (already referenced in context of pseudohypoparathyroidism)
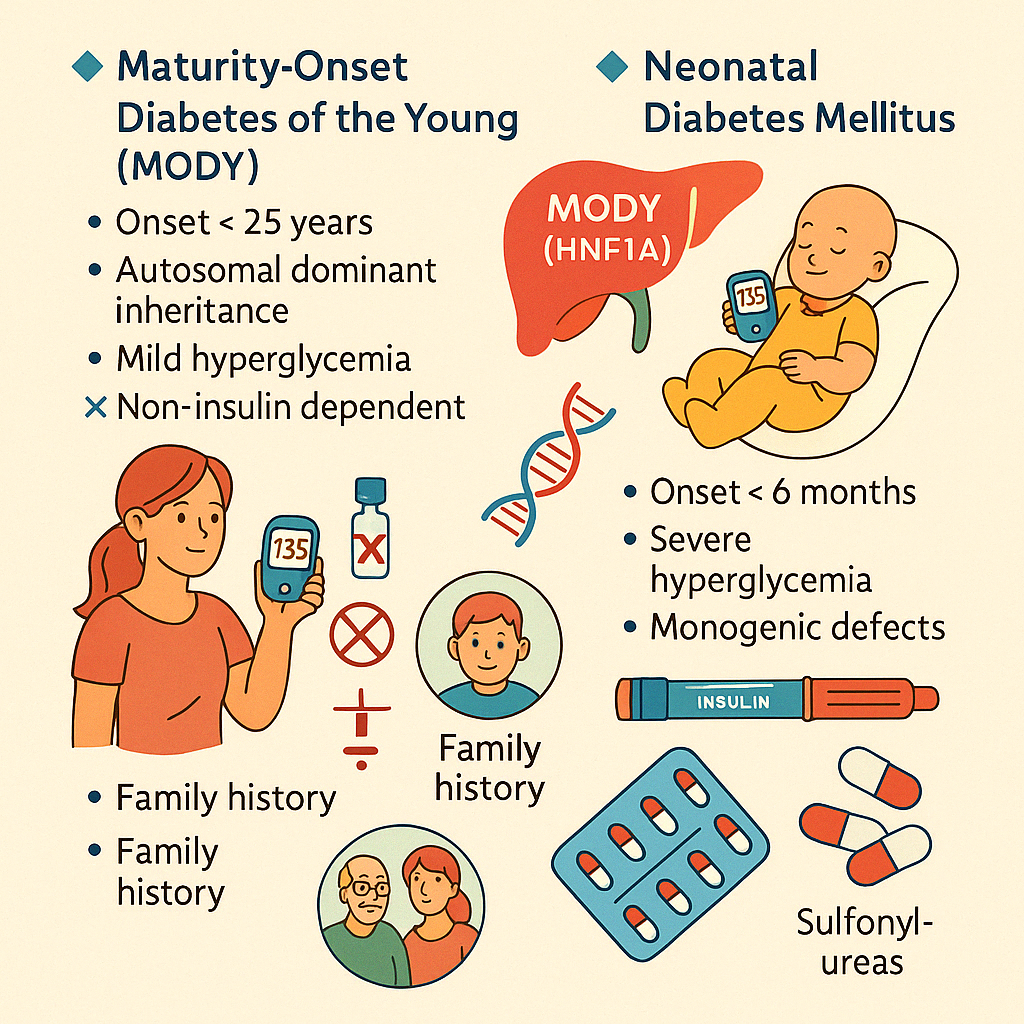
7. Genetic forms of Diabetes (e.g., MODY, neonatal diabetes)
Some rare forms of diabetes result from single-gene mutations and differ significantly from
type 1 or type 2 diabetes. Recognizing these monogenic diabetes syndromes allows for
targeted treatment and genetic counseling.
Maturity-Onset Diabetes of the Young (MODY)
MODY is an autosomal dominant form of diabetes caused by mutations in genes involved in β-
cell function. It typically presents before age 25 and is non-insulin dependent.
Key Features:
- Strong family history (autosomal dominant inheritance)
- Mild to moderate hyperglycemia
- Absence of obesity, insulin resistance, or ketosis
- Often misdiagnosed as type 1 or 2 diabetes
- Subtypes include HNF1A (MODY 3), HNF4A (MODY 1), GCK (MODY 2), etc.
Management:
- MODY 2: often requires no treatment
- MODY 3: very responsive to low-dose sulfonylureas
- MODY 1: may respond to oral agents, sometimes insulin
Neonatal Diabetes Mellitus (NDM)
NDM appears in the first 6 months of life and is due to monogenic defects in insulin
production. It may be transient or permanent.
Key Features:
- Severe hyperglycemia in infancy
- Failure to thrive
- May be associated with developmental delay or congenital anomalies
Causes:
- Mutations in KCNJ11, ABCC8, INS, and others
Management:
- Some forms respond to oral sulfonylureas (e.g., KCNJ11 mutations)
- Others require insulin therapy
- Genetic testing is crucial for diagnosis and guidance
Importance of Diagnosis:
Identifying monogenic diabetes:
- Avoids unnecessary insulin use
- Guides specific therapy
- Facilitates genetic couseling for family members