Disorders of Sexual Differentiation
This group of endocrine diseases includes the following conditions
- Congenital Adrenal Hyperplasia (CAH) – 21-hydroxylase deficiency (Already covered under Adrenal Diseases)
- Androgen Insensitivity Syndrome (AIS) – (Already covered under Adrenal Diseases)
- 5-Alpha Reductase Deficiency
- Gonadal Dysgenesis a. Complete Gonadal Dysgenesis (Swyer Syndrome) b. Mixed Gonadal Dysgenesis
- Ovotesticular DSD (True Hermaphroditism)
- 17β-Hydroxysteroid Dehydrogenase Deficiency
- Persistent Müllerian Duct Syndrome (PMDS)
1. Congenital Adrenal Hyperplasia (CAH) – 21-hydroxylase deficiency (Already covered under Adrenal Diseases)
2. Androgen Insensitivity Syndrome (AIS) – (Already covered under Adrenal Diseases)
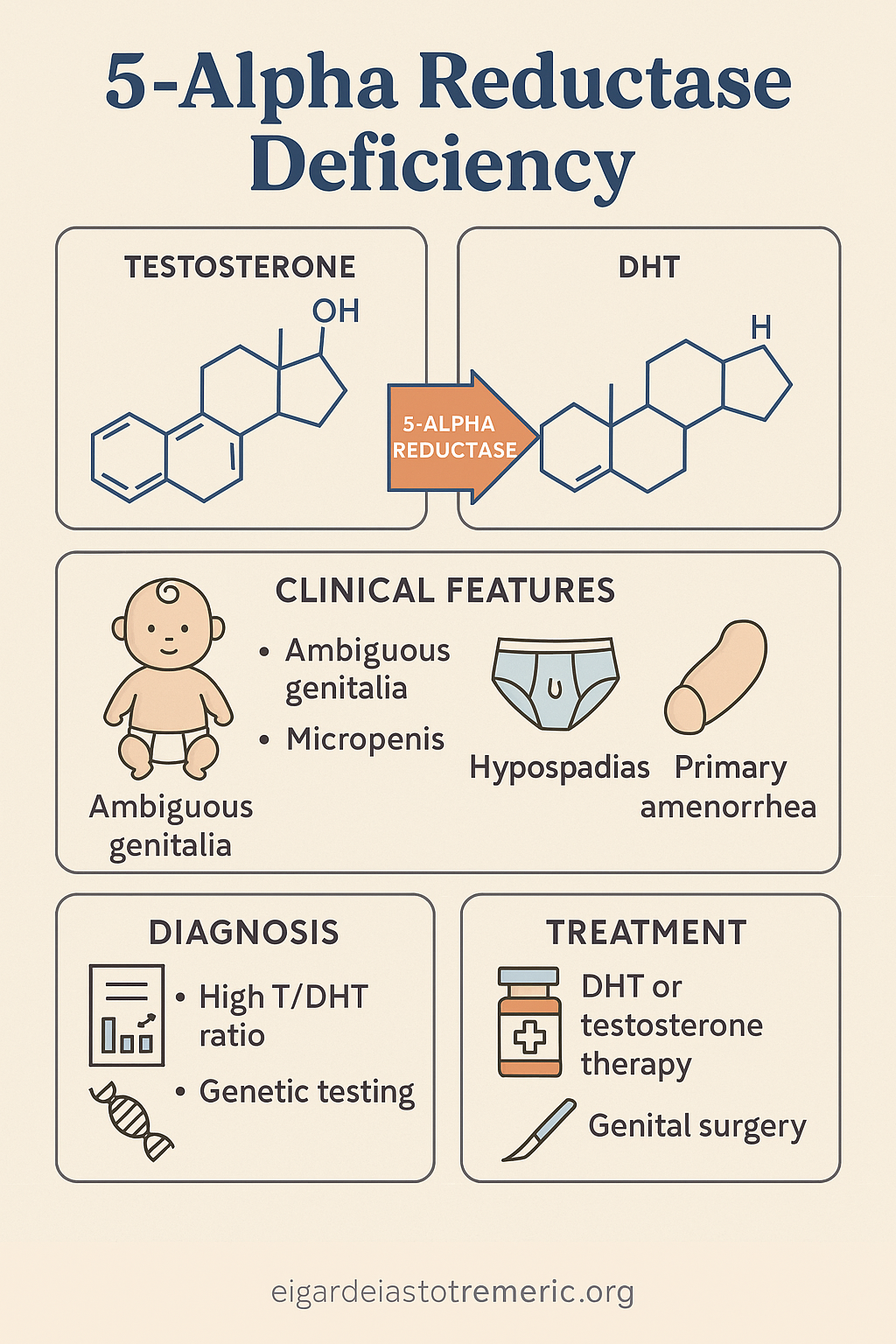
3. 5-Alpha Reductase Deficiency
Introduction
5-Alpha Reductase Deficiency is a rare autosomal recessive DSD caused by a mutation in the
SRD5A2 gene, which encodes the enzyme 5-alpha reductase type 2. This enzyme converts
testosterone to dihydrotestosterone (DHT), a crucial step for the development of external
male genitalia in utero.
Causes
- Genetic mutation in SRD5A2 gene
- Inheritance: autosomal recessive
- Affects 46,XY individuals who are genetically male but have undervirilized external genitalia
Clinical Manifestations
- At birth: ambiguous genitalia, ranging from female-like appearance to micropenis with hypospadias
- Testes are present, typically intra-abdominal or inguinal
- No uterus or fallopian tubes
- At puberty: increased muscle mass and voice deepening due to testosterone; however, minimal or absent facial/body hair and no significant phallic growth
- Some individuals adopt male gender role at puberty due to virilization
Diagnosis
- Karyotype: 46,XY
- Normal or elevated testosterone, low DHT, high T/DHT ratio
- Ultrasound: absence of uterus; presence of testes
- Genetic testing confirms SRD5A2 mutation
Management
- Gender assignment counseling is central and individualized
- Surgical correction (e.g., hypospadias repair or gonadectomy depending on gender identity)
- Hormonal therapy: DHT gel or testosterone if male gender is chosen
- Psychosocial support throughout adolescence and adulthood
Complications
- Infertility is common
- Risk of gonadal malignancy if undescended testes are retained
- Psychosexual challenges without appropriate counselling
4. Gonadal Dysgenesis
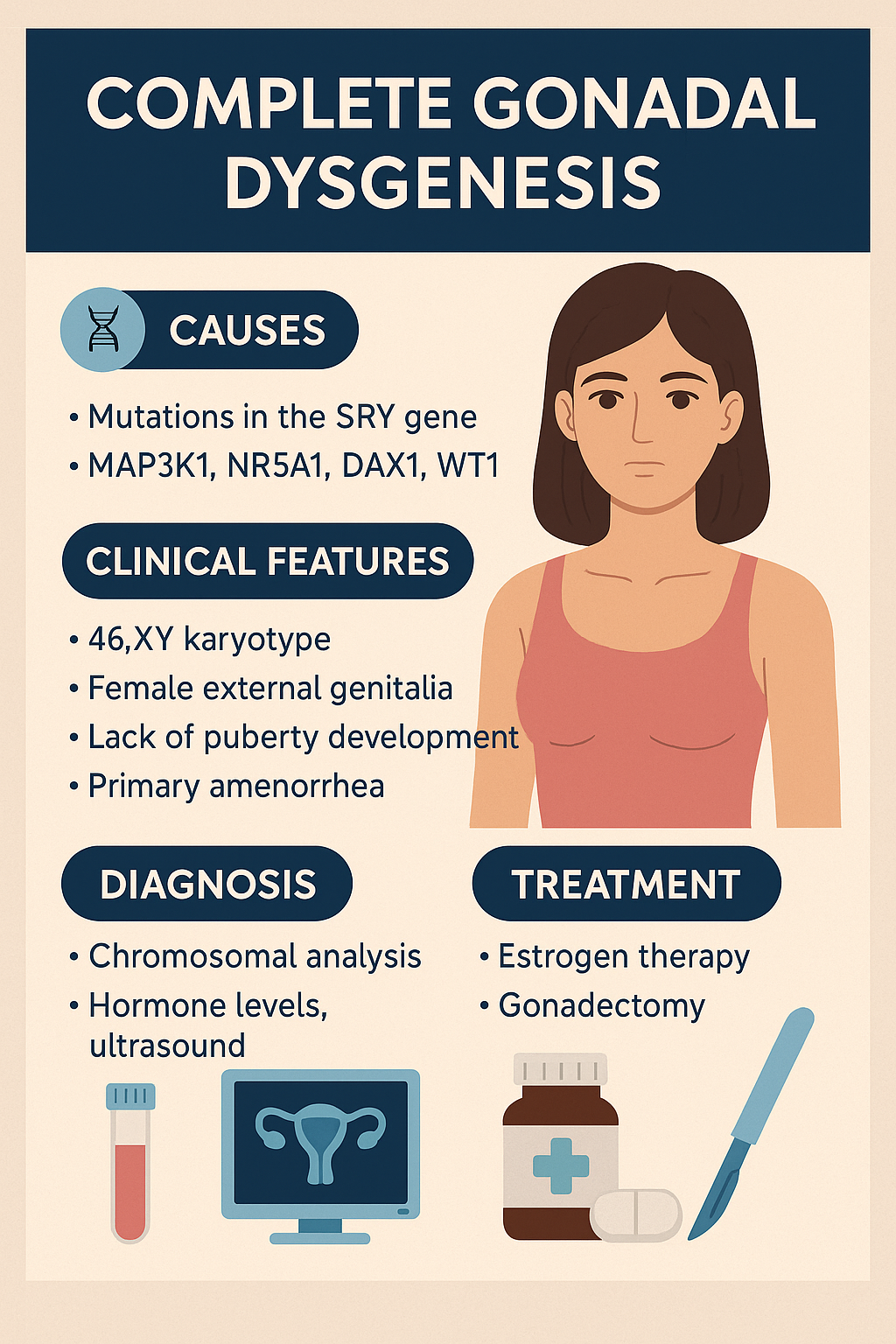
Complete Gonadal Dysgenesis (Swyer Syndrome)
Introduction
Swyer Syndrome, also called Complete Gonadal Dysgenesis, is a rare DSD in individuals with
a 46,XY karyotype who develop normal female external genitalia but have nonfunctional,
fibrous streak gonads instead of testes or ovaries. It results from failure of testis
differentiation despite the presence of the SRY gene or downstream signaling defects.
Causes
- Mutations in the SRY gene (sex-determining region on Y chromosome)
- Can also involve other genes: MAP3K1, NR5A1, DAX1, WT1
- Typically sporadic, but familial forms exist
Clinical Manifestations
- Normal female external genitalia
- Normal Müllerian structures (uterus, fallopian tubes, vagina)
- No breast development or puberty (due to lack of estrogen)
- Primary amenorrhea is often the first clue
- Tall stature may be present
- No secondary sexual hair or minimal pubic/axillary hair
- Gonads are nonfunctional streaks, at risk of malignancy (gonadoblastoma)
Diagnosis
- Karyotype: 46,XY
- Low estrogen, high FSH and LH (hypergonadotropic hypogonadism)
- Ultrasound or MRI: uterus present, small or absent gonads
- SRY gene testing or broader DSD genetic panel
- Biopsy may confirm dysgenetic gonads
Management
- Gonadectomy is essential to prevent malignancy
- Estrogen replacement therapy for breast development and menstruation
- Later, progestins added to induce regular cycles
- Fertility with donor oocytes is possible
- Ongoing psychosocial support and genetic counseling
Complications
- Gonadal tumors (esp. gonadoblastoma, dysgerminoma)
- Psychological stress without proper education/support
- Infertility, though pregnancy is possible with assisted reproduction
Mixed Gonadal Dysgenesis
Introduction
Mixed Gonadal Dysgenesis (MGD) is a form of DSD characterized by asymmetrical gonadal
development, typically with a testis on one side and a streak gonad on the other. It occurs
in individuals with mosaic karyotypes, most commonly 45,X/46,XY. The phenotype can vary
widely — from near-normal male to ambiguous or predominantly female genitalia.
Causes
- Chromosomal mosaicism, most often 45,X/46,XY
- Abnormalities in sex chromosome differentiation
- Resulting in incomplete or asymmetric testicular development
Clinical Manifestations
- Ambiguous genitalia at birth (most common presentation)
- One palpable testis; contralateral gonad often nonpalpable (streak)
- Presence of Müllerian structures (fallopian tube, uterus) on the streak side
- May present with hypospadias, undescended testis, or clitoromegaly
- Short stature (due to X monosomy)
- Variable breast development and menstruation if raised female
- Risk of gonadal tumors, particularly gonadoblastoma or dysgerminoma
Diagnosis
- Karyotype: mosaic (e.g., 45,X/46,XY)
- Ultrasound or MRI: detect Müllerian structures and gonads
- Hormonal profile: may show low testosterone or discordant hormone levels
- Laparoscopy and biopsy for gonadal characterization
Management
- Individualized gender assignment based on phenotype, internal anatomy, and family/cultural context
- Prophylactic removal of streak gonad due to tumor risk
- Hormonal therapy based on chosen gender
- Surgical correction of ambiguous genitalia
- Psychosocial support and long-term follow-up
Complications
- Malignancy risk (gonadoblastoma > dysgerminoma)
- Infertility (especially in females)
- Psychological challenges during puberty and identity formation
Engage With Dr. Vipin Mishra
Whether you are dealing with chronic endocrine condition or just want to understand your body better, or wish to obtain an exalted consciousness, you can take help and guidance from Dr. Vipin Mishra.
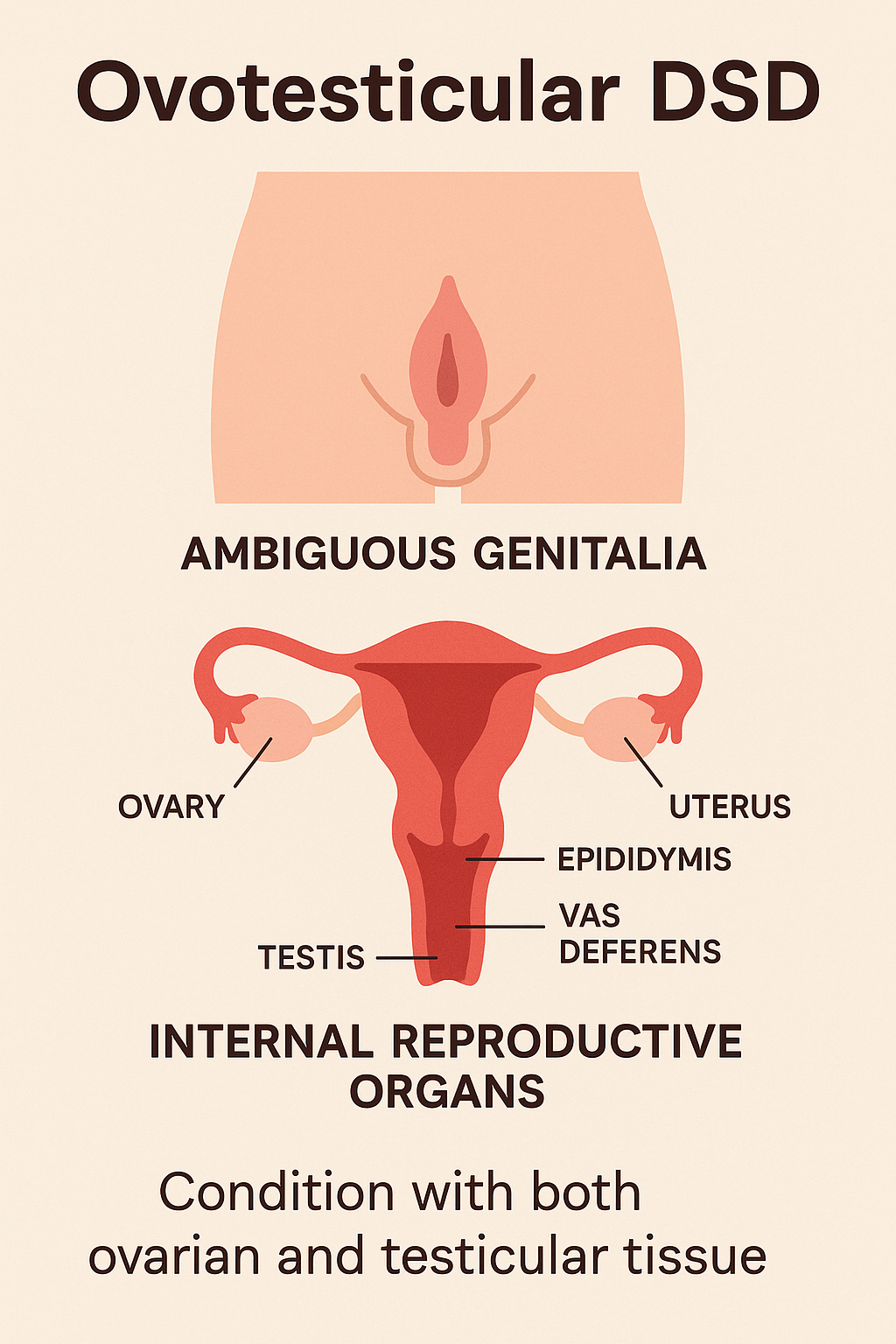
Engage With Dr. Vipin Mishra5. Ovotesticular DSD (True Hermaphroditism)
Introduction
Ovotesticular DSD, formerly known as true hermaphroditism, is a condition where
individuals have both ovarian and testicular tissue. This may be present as ovotestis
(combined tissue) or as one ovary and one testis. It is an extremely rare form of DSD and
can occur in individuals with 46,XX, 46,XY, or mosaic karyotypes.
Causes
- Exact cause is often unknown
- In some cases, may involve SRY translocation onto the X chromosome
- Chromosomal variations: most common is 46,XX, followed by mosaic forms (e.g., 46,XX/46,XY)
Clinical Manifestations
- Ambiguous genitalia at birth
- Varying degrees of genital virilization or feminization
- Presence of a uterus and/or vagina in many cases
- Breast development and menstruation may occur at puberty in some XX individuals
- Gonads may include:
- One ovary and one testis
- Two ovotestes
- One ovotestis and one streak gonad
Diagnosis
- Karyotype: often 46,XX; but 46,XY or mosaicism also possible
- Imaging: pelvic ultrasound or MRI for internal organs
- Hormonal assays: variable depending on the dominant gonadal tissue
- Laparoscopy and biopsy for definitive diagnosis of gonadal histology
Management
- Gender assignment based on genital anatomy, gonadal function, and parental preference
- Surgical correction to align physical features with chosen gender
- Removal of dysgenetic gonads to reduce malignancy risk
- Hormone replacement therapy as needed
- Ongoing psychosocial support and multidisciplinary care
Complications
- Infertility (though rare cases of fertility have been documented)
- Risk of gonadal tumors, especially in dysgenetic gonads
- Psychosexual stress without adequate counseling and education
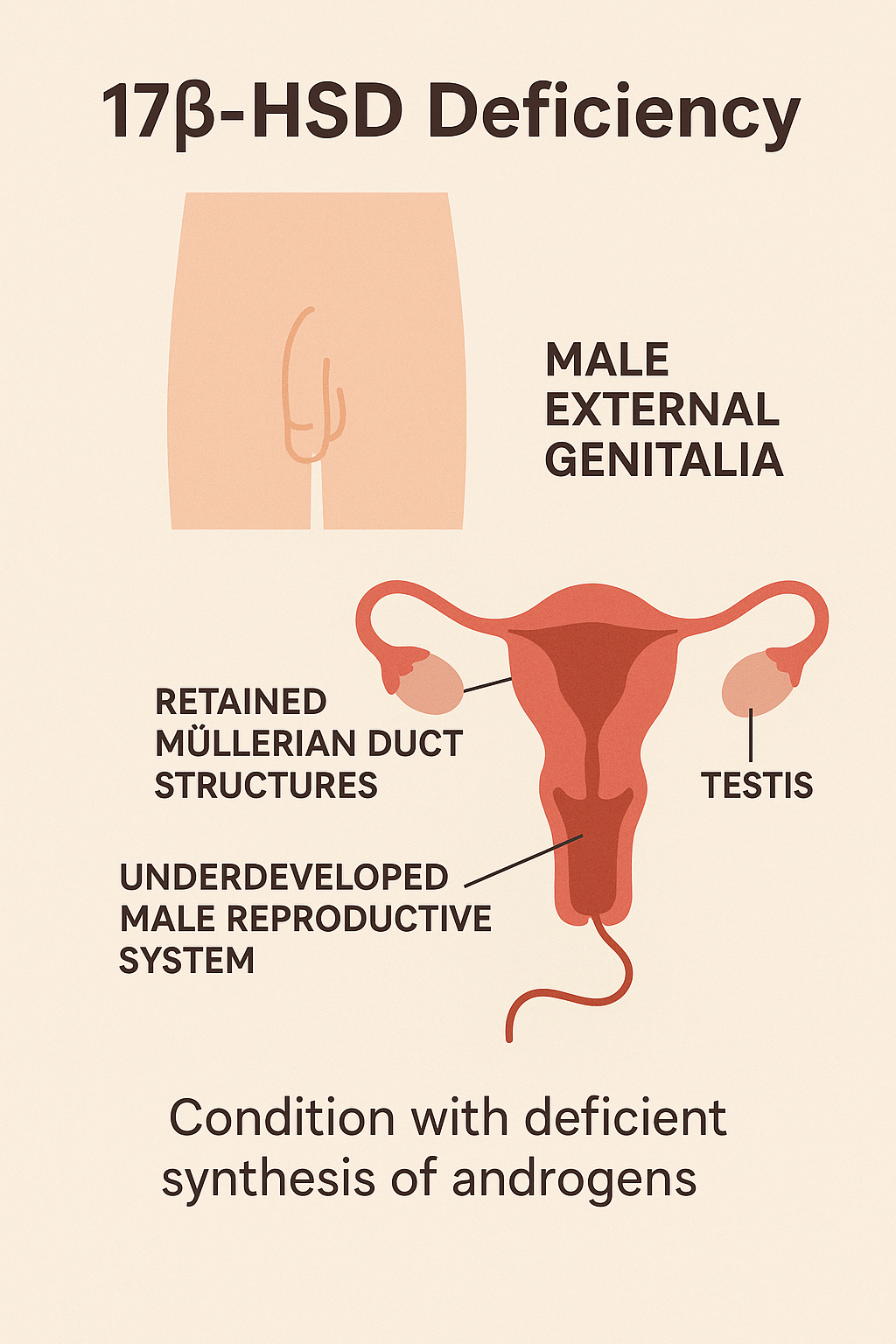
6. 17β-Hydroxysteroid Dehydrogenase Deficiency
Introduction
17β-Hydroxysteroid Dehydrogenase Deficiency is a rare autosomal recessive disorder
affecting sex steroid metabolism. It leads to impaired conversion of androstenedione to
testosterone in males, resulting in undervirilization of individuals with a 46,XY karyotype. It
may also affect estrogen production, impacting females.
Causes
- Mutation in the HSD17B3 gene, coding for the type 3 isoenzyme of 17β-HSD
- Autosomal recessive inheritance
- Affects the final steps of testosterone biosynthesis in the testes
Clinical Manifestations
- 46,XY individuals:
- Female-appearing or ambiguous external genitalia at birth
- Small phallus or clitoromegaly
- Inguinal or labial masses (testes)
- No Müllerian structures (uterus, fallopian tubes absent)
- At puberty:
- Increased testosterone production from other pathways may cause spontaneous virilization (deep voice, clitoromegaly, muscle mass increase)
- 46,XX individuals: generally unaffected
Diagnosis
- Karyotype: 46,XY
- Hormonal profile:
- Elevated androstenedione
- Low or inappropriately normal testosterone
- High androstenedione/testosterone ratio
- Genetic testing for HSD17B3 mutation
- Imaging: undescended testes, no uterus
Management
- Gender assignment should be individualized after multidisciplinary evaluation
- Surgical gonadectomy is advised if female gender is assigned (due to malignancy risk)
- Testosterone therapy for males who wish to enhance masculinization
- Surgical correction of external genitalia (e.g., hypospadias repair, genital reconstruction)
- Psychosocial counseling throughout life
Complications
- Gonadal tumors in undescended testes if not removed
- Infertility is common
- Psychosexual adjustment issues if counseling is inadequate
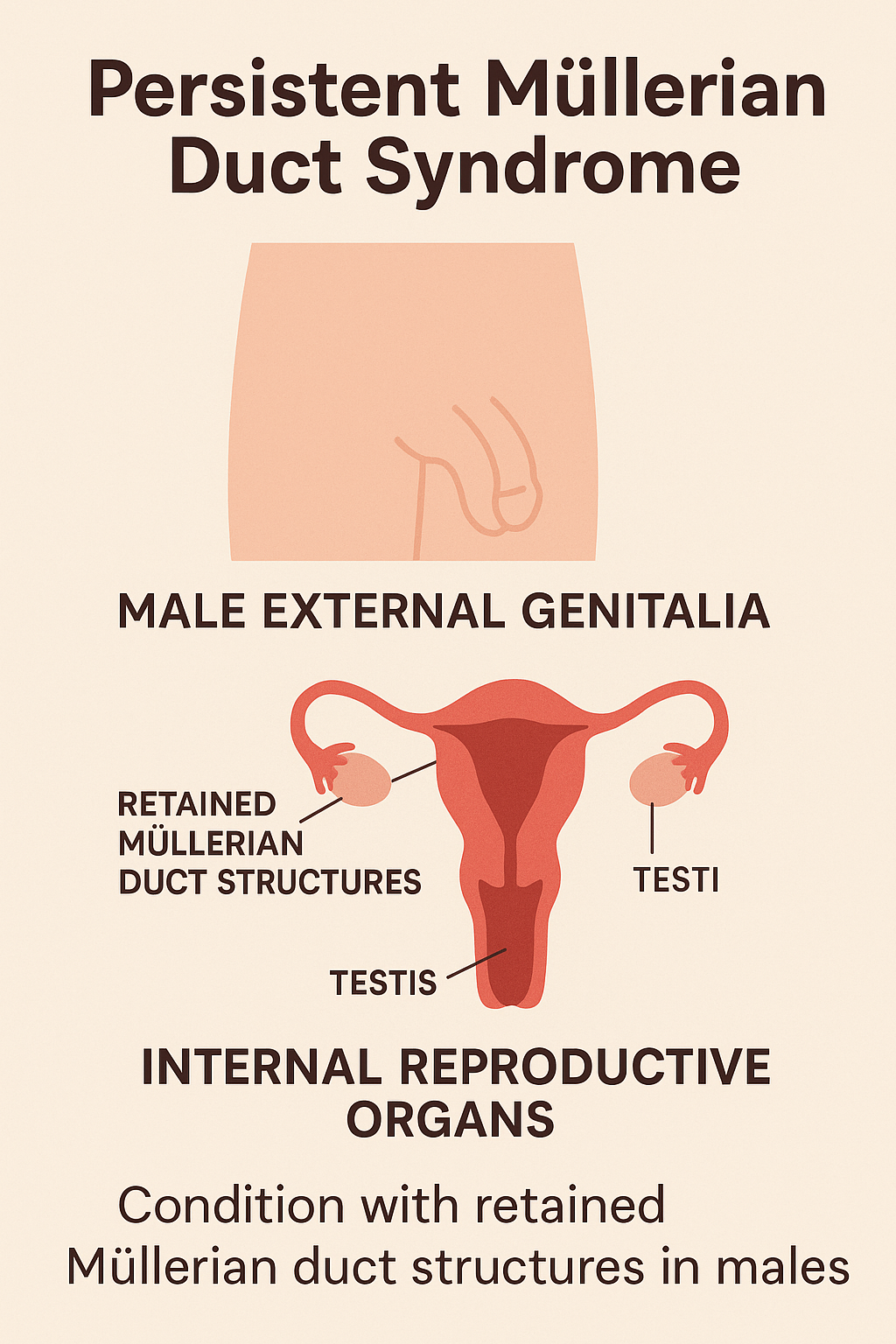
7. Persistent Müllerian Duct Syndrome (PMDS)
Introduction
Persistent Müllerian Duct Syndrome (PMDS) is a rare DSD where genetically male
individuals (46,XY) retain Müllerian duct structures (uterus, fallopian tubes) due to failure
of regression during embryogenesis. Despite having normal male external genitalia, these
individuals possess internal female reproductive structures.
Causes
- Mutation in the AMH gene (Anti-Müllerian Hormone) or its receptor gene (AMHR2)
- Inheritance: autosomal recessive
- Deficiency or insensitivity to AMH, which is responsible for Müllerian duct regression in males
Clinical Manifestations
- Typically diagnosed during hernia or cryptorchidism surgery in childhood
- Normal male external genitalia
- Undescended testes (unilateral or bilateral)
- Uterus and fallopian tubes found incidentally
- Rarely presents with infertility in adulthood
Diagnosis
- Karyotype: 46,XY
- Normal testosterone and male secondary sexual characteristics
- Pelvic imaging (US/MRI) reveals uterus or Müllerian remnants
- Hormonal levels typically normal for a male
- Genetic testing for AMH or AMHR2 mutations
Management
- Surgical removal of Müllerian structures to prevent obstruction or malignancy
- Orchidopexy or orchiectomy depending on testicular location and function
- Preservation of fertility when possible
- Regular follow-up for malignancy surveillance
Complications
- Infertility due to undescended or dysplastic testes
- Inguinal hernia recurrence if Müllerian structures are not addressed
- Malignancy risk in retained Müllerian or gonadal tissue
- Psychological concerns if discovered late without prior counseling