Endocrine Tumors and Syndromes
This group of endocrine diseases includes the following conditions
- Multiple Endocrine Neoplasia Type 1 (MEN1)
- Multiple Endocrine Neoplasia Type 2 (MEN2A and MEN2B)
- Pheochromocytoma (also covered under adrenal disorders)
- Neuroendocrine Tumors (NETs) — GI and pancreatic
- Carcinoid Syndrome
- Paraneoplastic Endocrine Syndromes
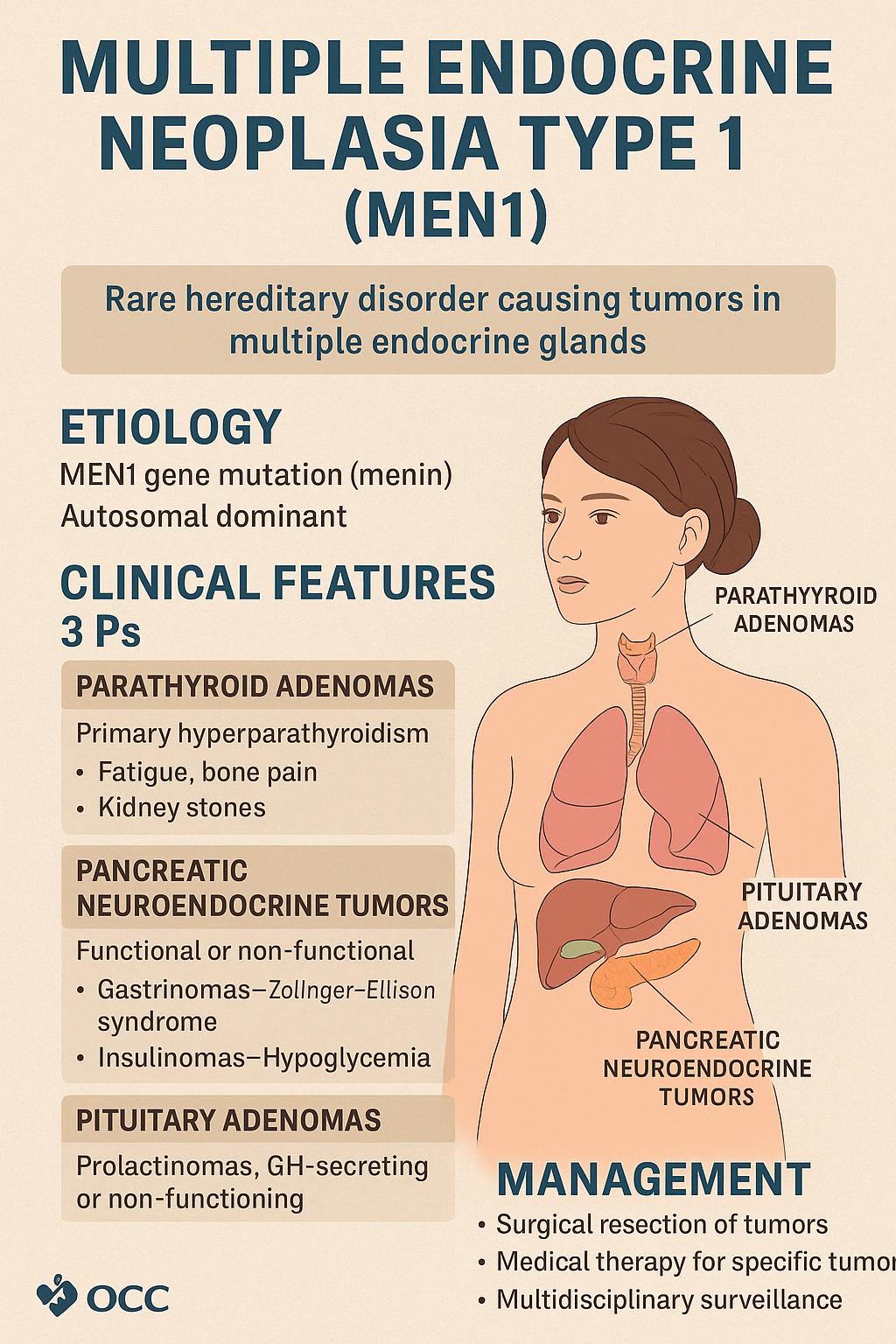
1. Multiple Endocrine Neoplasia Type 1 (MEN1)
MEN1 is a rare, inherited autosomal dominant syndrome caused by mutations in the MEN1 gene (which encodes menin, a tumor suppressor protein). It leads to the development of multiple endocrine and non-endocrine tumors, typically involving the "3 Ps":
- Parathyroid adenomas (most common)
- Pancreatic neuroendocrine tumors
- Pituitary adenomas
Clinical Features
- Parathyroid Adenomas (95%)
- Present with primary hyperparathyroidism
- Symptoms: fatigue, bone pain, kidney stones, hypercalcemia
- Pancreatic Neuroendocrine Tumors (~60%)
- Can be functional or non-functional
- Gastrinomas → Zollinger–Ellison syndrome (severe peptic ulcers, diarrhea)
- Insulinomas → hypoglycemia
- Glucagonomas, VIPomas, somatostatinomas (rare)
- Pituitary Adenomas (~40%)
- Prolactinomas, GH-secreting tumors, or non-functioning adenomas
- Symptoms: headache, visual disturbances, menstrual dysfunction, acromegaly
Other associated tumors may include:
- Adrenal tumors
- Bronchial or thymic carcinoids
- Lipomas and angiofibromas (non-endocrine)
Diagnosis
- Based on clinical presentation and family history
- Genetic testing for MEN1 mutation
- Calcium and PTH, hormonal work-up, MRI/CT imaging of pituitary, pancreas, and chest
- Annual surveillance in known carriers
Management
- Surgical resection of symptomatic tumors
- Medical therapy: dopamine agonists for prolactinomas, PPIs for gastrinomas
- Lifelong multidisciplinary surveillance for early detection and treatment
Early diagnosis in at-risk individuals is key to improving quality of life and longevit.
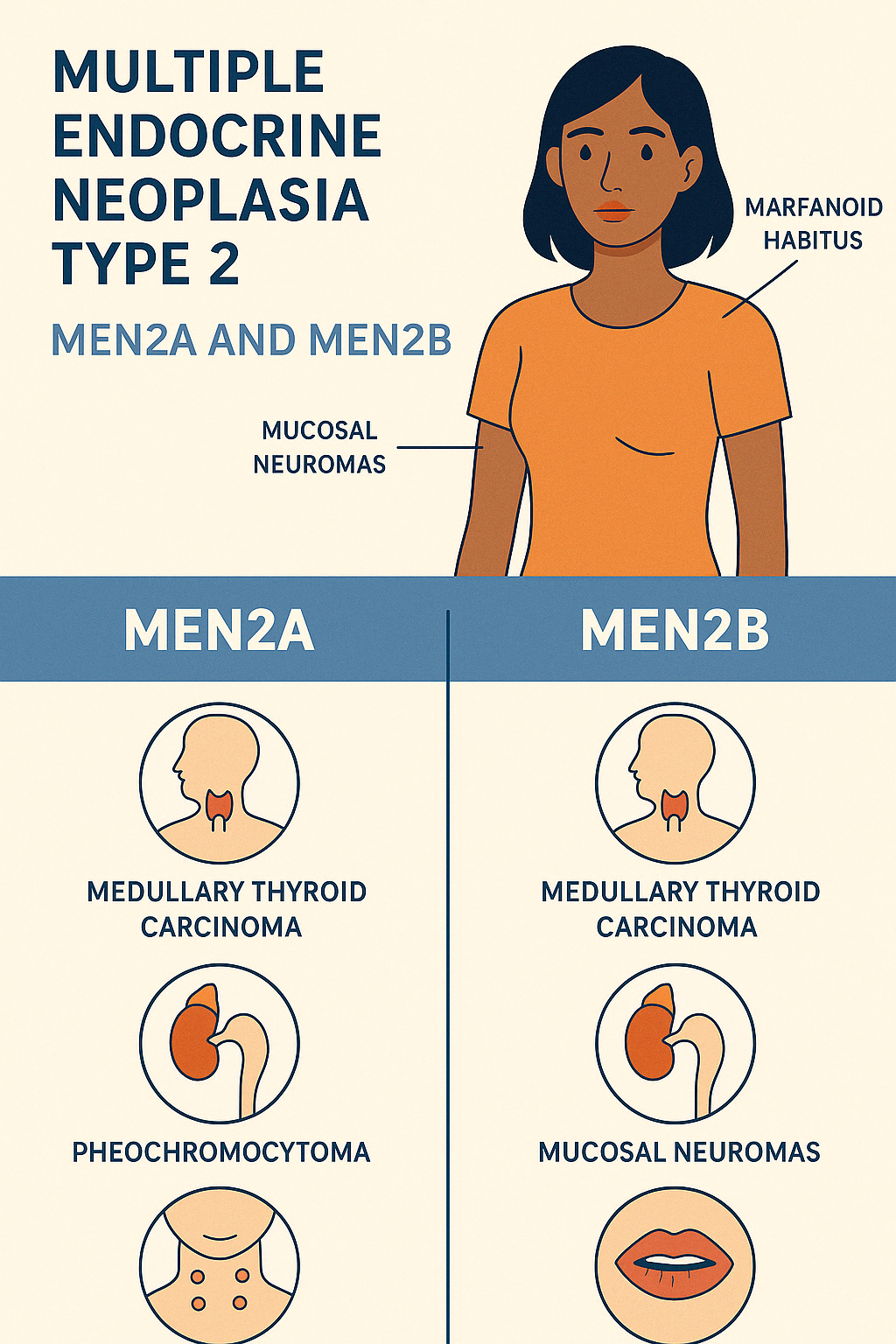
2. Multiple Endocrine Neoplasia Type 2 (MEN2A and MEN2B)
MEN2 is a rare autosomal dominant syndrome caused by mutations in the RET proto-oncogene. It is subdivided into MEN2A, MEN2B, and Familial Medullary Thyroid Carcinoma (FMTC) — all with a high risk of developing medullary thyroid carcinoma (MTC).
MEN2A ("Classical MEN2") – 90–95% of MEN2 cases
Core Triad:
- Medullary Thyroid Carcinoma (MTC) – nearly 100% of cases
- Pheochromocytoma – ~50%
- Primary Hyperparathyroidism – ~25%
MEN2B (Previously MEN3) – 5–10% of MEN2 cases
Aggressive variant presenting earlier in life
- Medullary Thyroid Carcinoma (more aggressive)
- Pheochromocytoma
- Mucosal neuromas (lips, tongue, eyelids)
- Marfanoid habitus
- GI symptoms (ganglioneuromatosis → constipation or diarrhea)
Clinical Features
- MTC: early metastasis, calcitonin-producing tumor
- Pheochromocytoma: episodic hypertension, palpitations, headaches
- Hyperparathyroidism (MEN2A): hypercalcemia, kidney stones
- Neuromas & marfanoid body (MEN2B): tall, slender limbs, joint laxity
Diagnosis
- Genetic testing for RET mutations (definitive)
- Serum calcitonin, plasma metanephrines, calcium & PTH
- Imaging: neck US, CT/MRI for pheochromocytoma
Management
- Prophylactic thyroidectomy in early childhood (especially in MEN2B)
- Surgical removal of pheochromocytoma
- Monitoring and management of parathyroid disease
- Genetic counseling and screening of family members
Early detection through genetic screening dramatically improves outcomes, especially by preventing MTC progression.
3. Pheochromocytoma (covered already under adrenal disorders) Pheochromocytoma — (Detailed under the section of Adrenal Diseases. / Link)
Engage With Dr. Vipin Mishra
Whether you are dealing with chronic endocrine condition or just want to understand your body better, or wish to obtain an exalted consciousness, you can take help and guidance from Dr. Vipin Mishra.
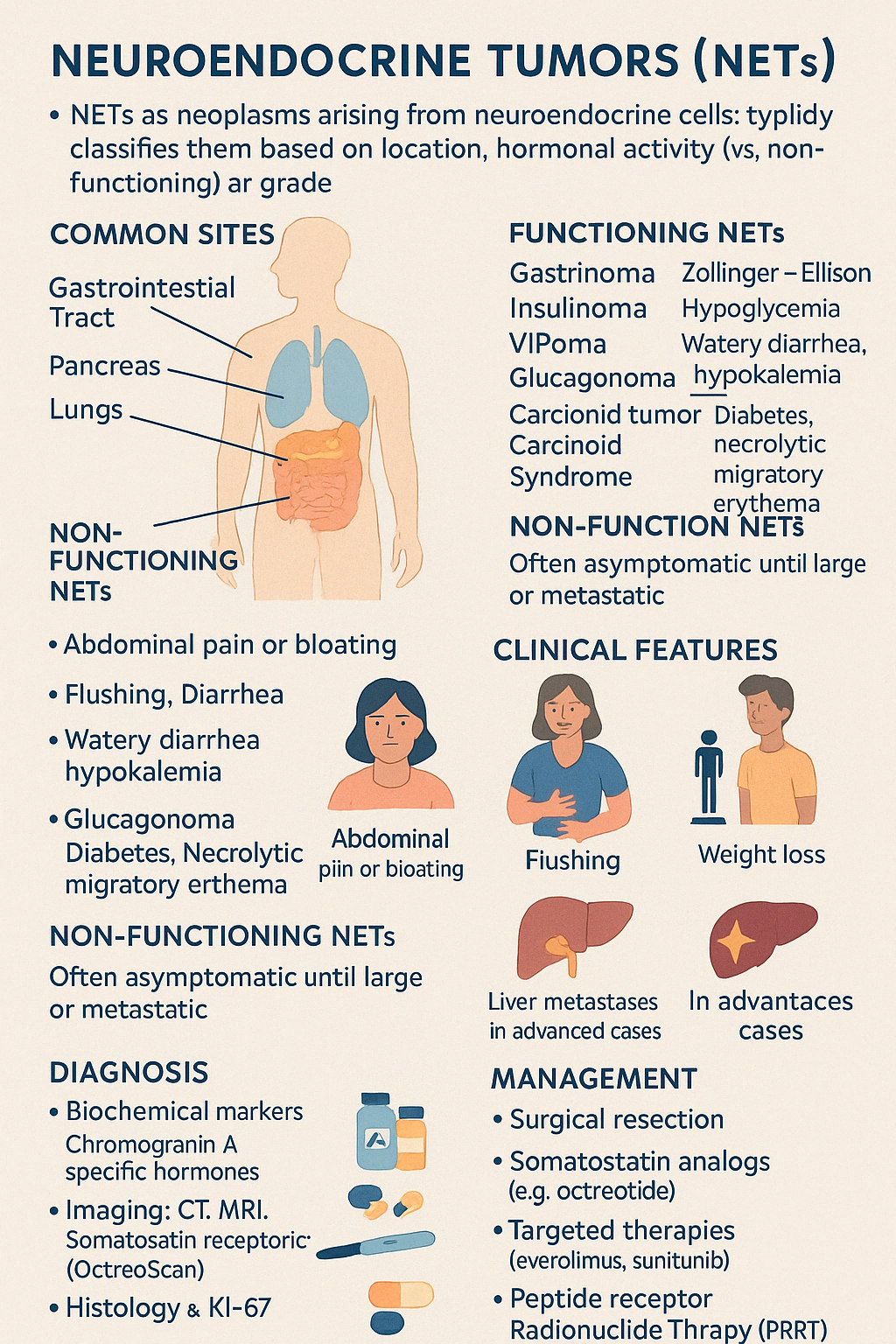
Engage With Dr. Vipin Mishra4. Neuroendocrine Tumors (NETs) — GI and pancreatic
Neuroendocrine Tumors (NETs) are a diverse group of neoplasms arising from neuroendocrine cells, which have both neuronal and endocrine properties. These tumors can secrete hormones and bioactive amines, and may be found throughout the body — most commonly in the gastrointestinal tract, pancreas, and lungs.
They are classified based on:
- Location (e.g., pancreatic, intestinal, bronchial)
- Hormonal activity (functioning vs. non-functioning)
- Grade (based on mitotic count and Ki-67 index)
Functioning NETs (produce hormones):
- Gastrinoma → Zollinger–Ellison Syndrome
- Insulinoma → Hypoglycemia
- VIPoma → Watery diarrhea, hypokalemia
- Glucagonoma → Diabetes, necrolytic migratory erythema
- Carcinoid Tumor → Carcinoid Syndrome (see next topic)
Non-functioning NETs:
- Often asymptomatic until large or metastatic
- Symptoms arise from mass effect or incidental detection
Clinical Features:
- Abdominal pain or bloating
- Flushing, diarrhea (in functioning NETs)
- Weight loss, peptic ulcers (gastrinomas)
- Hypoglycemia (insulinomas)
- Liver metastases in advanced cases
Diagnosis:
- Biochemical markers: Chromogranin A, specific hormones (insulin, gastrin, glucagon, VIP)
- Imaging: CT, MRI, somatostatin receptor scintigraphy (OctreoScan), PET-DOTATATE
- Histology and Ki-67 for tumor grade
Management:
- Surgical resection (curative in localized tumors)
- Somatostatin analogs (e.g., octreotide) to control hormone secretion and slow tumor growth
- Targeted therapies: everolimus, sunitinib
- Peptide Receptor Radionuclide Therapy (PRRT)
- Chemotherapy for high-grade tumors
NETs often progress slowly, and early detection offers a chance for long-term control or cure.
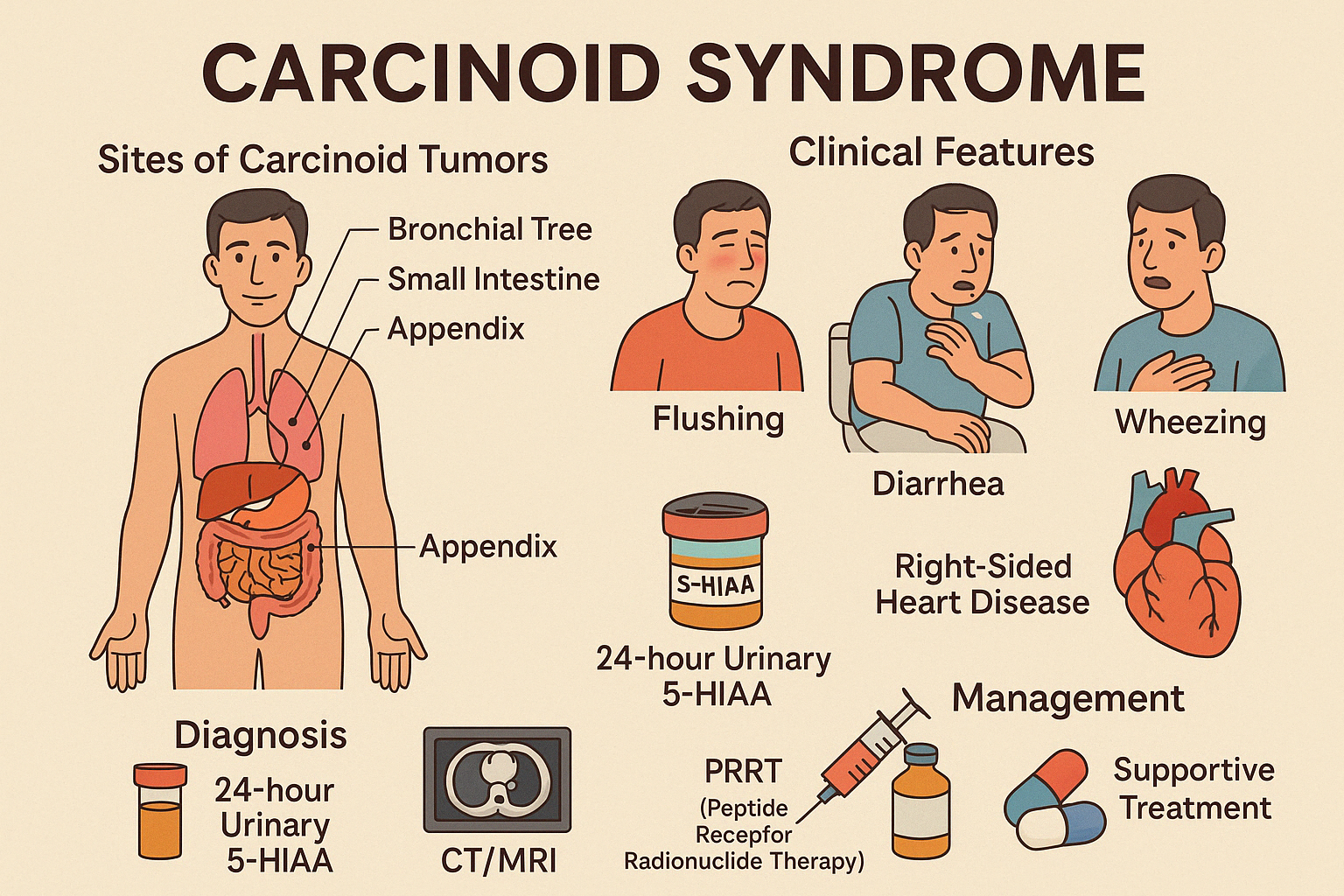
5. Carcinoid Syndrome
Carcinoid Syndrome is a clinical condition caused by hormone-secreting neuroendocrine tumors, primarily serotonin, along with histamine, bradykinin, and prostaglandins. It typically occurs when carcinoid tumors metastasize to the liver, bypassing hepatic degradation of these bioactive substances.
Most common sites of carcinoid tumors:
- Small intestine (ileum)
- Appendix
- Bronchial tree (lungs)
Clinical Features:
- Flushing (episodic, often facial)
- Diarrhea (secretory, watery)
- Wheezing or bronchospasm
- Right-sided heart disease: tricuspid and pulmonary valve fibrosis (unique to this syndrome)
- Abdominal pain or intestinal obstruction (mass effect)
Diagnosis:
- 24-hour urinary 5-HIAA (serotonin metabolite) — hallmark test
- Serum Chromogranin A
- CT/MRI, OctreoScan, or PET-DOTATATE for localization
- Echocardiography if heart involvement suspected
Management:
- Somatostatin analogs (e.g., octreotide, lanreotide) to control symptoms and tumor growth
- Surgical debulking if feasible
- PRRT (Peptide Receptor Radionuclide Therapy) in metastatic or inoperable cases
- Supportive treatment for diarrhea, bronchospasm, or cardiac involvement
Prompt recognition and targeted therapy can improve both quality of life and long-term outcomes.
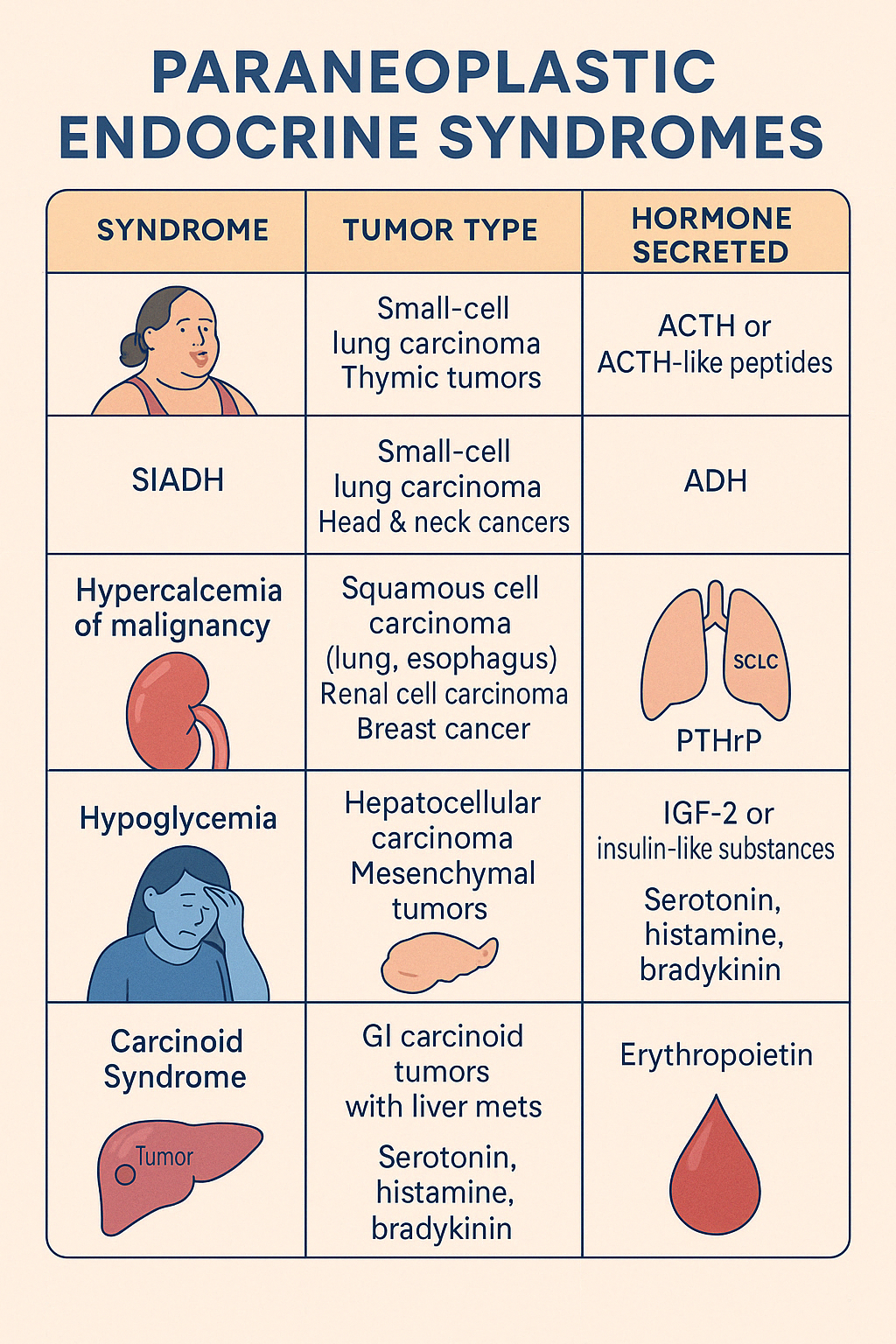
6. Paraneoplastic Endocrine Syndromes
Paraneoplastic Endocrine Syndromes occur when non-endocrine tumors produce hormones or hormone-like substances, leading to systemic endocrine effects not directly related to tumor invasion. These syndromes are often the first clue to an occult malignancy.
Common Paraneoplastic Hormonal Syndromes:
| Syndrome | Tumor Type | Hormone Secreted |
|---|---|---|
| Cushing's Syndrome | Small-cell lung carcinoma (SCLC), thymic tumors | ACTH or ACTH-like peptides |
| SIADH | SCLC, head & neck cancers | ADH |
| Hypercalcemia of malignancy | Squamous cell carcinoma (lung, esophagus), renal cell carcinoma, breast cancer | PTHrP |
| Hypoglycemia | Hepatocellular carcinoma, mesenchymal tumors | IGF-2 or insulin-like substances |
| Carcinoid Syndrome | GI carcinoid tumors with liver mets | Serotonin, histamine, bradykinin |
| Erythrocytosis | Renal cell carcinoma, hepatoma | Erythropoietin |
Clinical Features:
- Cushingoid features, hyponatremia, hypercalcemia, hypoglycemia, or flushing/diarrhea depending on the hormone
- Often systemic and severe in advanced disease
- Symptoms may precede tumor detection
Diagnosis:
- Hormonal assays (ACTH, ADH, PTHrP, IGF-2, etc.)
- Imaging to locate the tumor (CT, PET, MRI)
- Tumor markers and biopsy when needed
Management:
- Treat the underlying malignancy (surgery, chemo, radiation)
- Symptomatic hormonal management (e.g., ketoconazole for hypercortisolism, bisphosphonates for hypercalcemia, somatostatin analogs for carcinoid syndrome)
- Supportive care for complications
Recognizing paraneoplastic syndromes enables earlier diagnosis and treatment of hidden cancers, often improving patient outcomes.